Proteome and Phosphoproteome Profiling Reveal the Toxic Mechanism of Clostridium perfringens Epsilon Toxin in MDCK Cells


Abstract

1. Introduction
2. Results
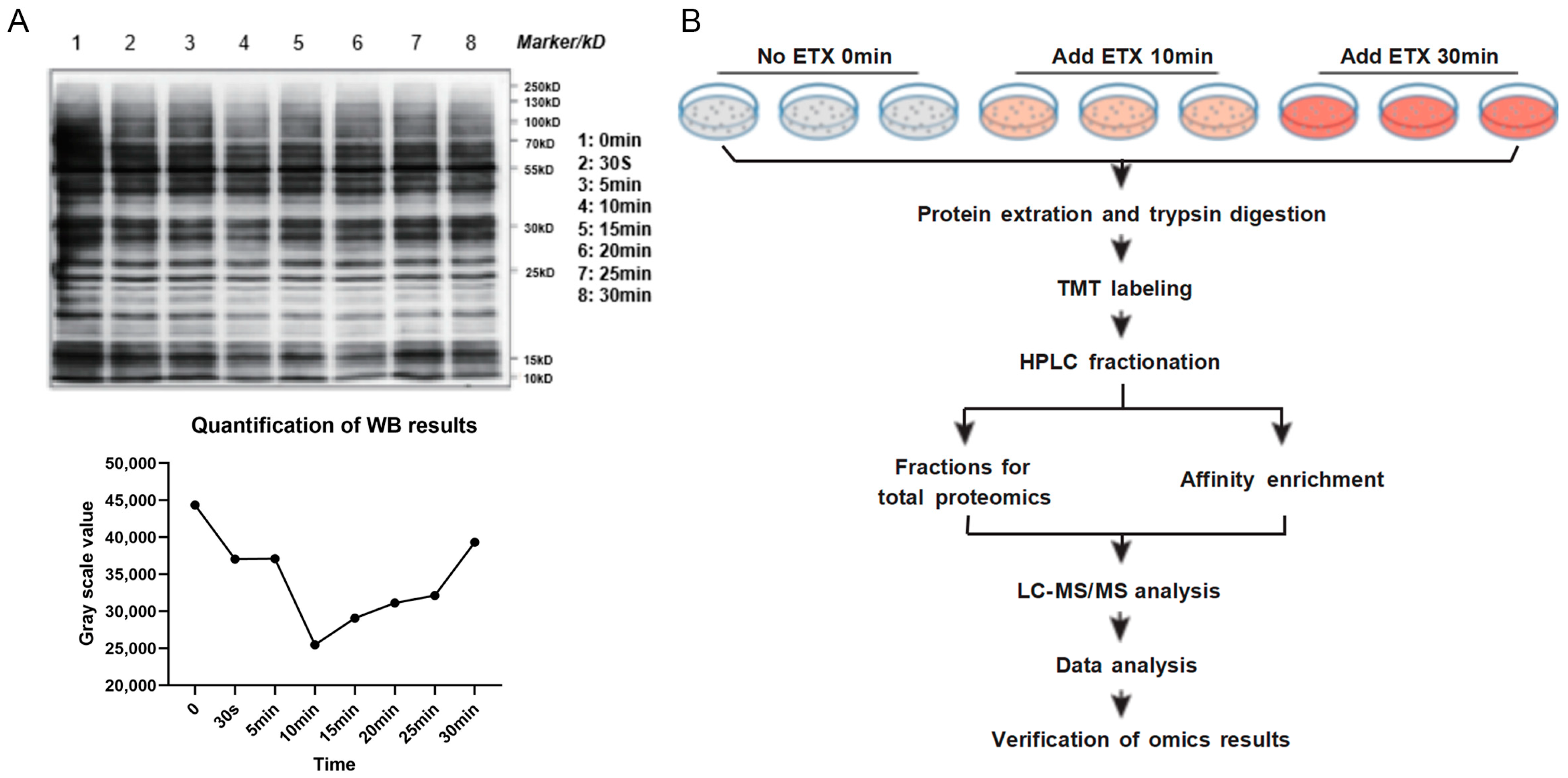
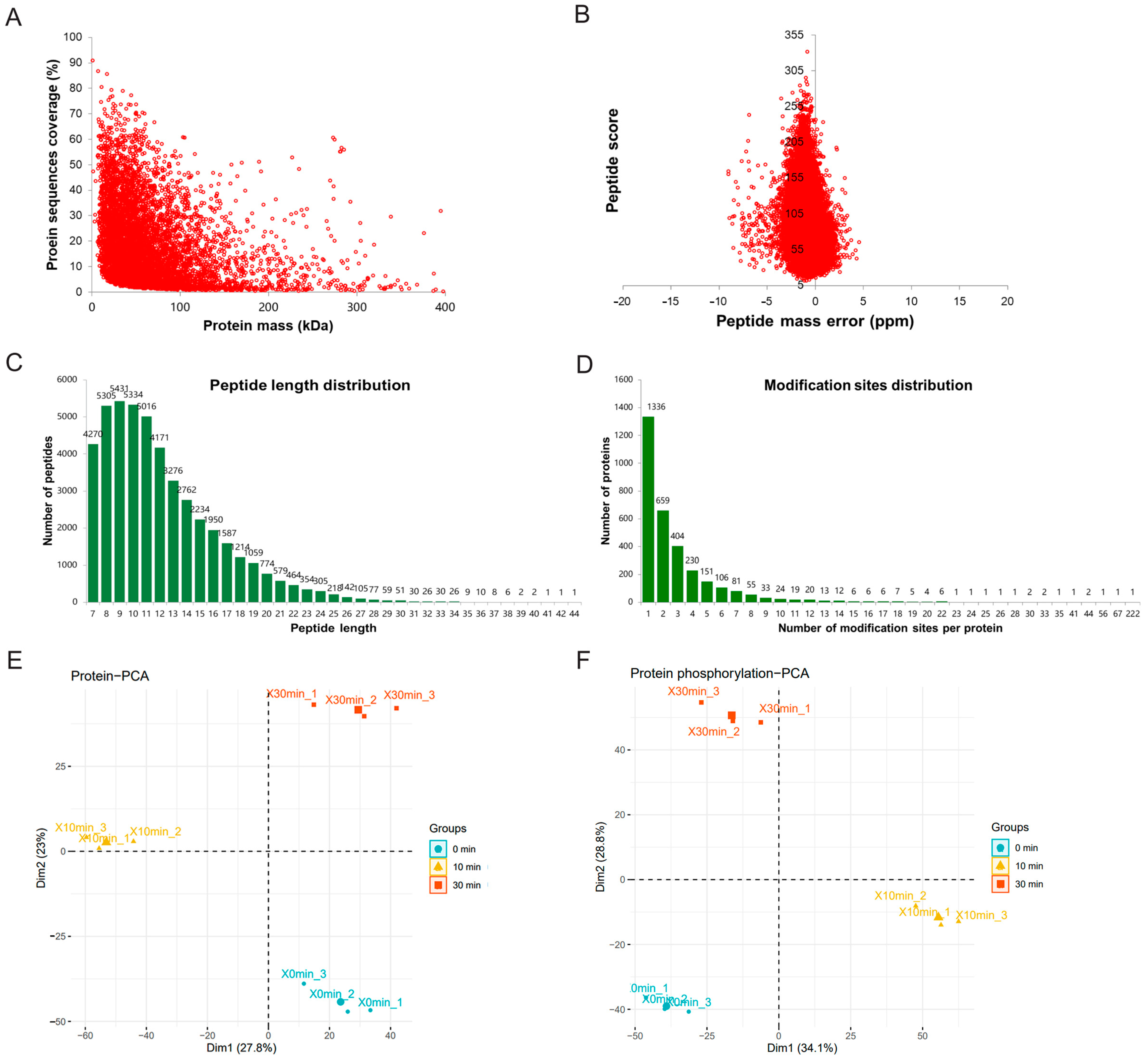
2.1. Pre-Assessment of Phosphorylation on MDCK Cells and Workflow
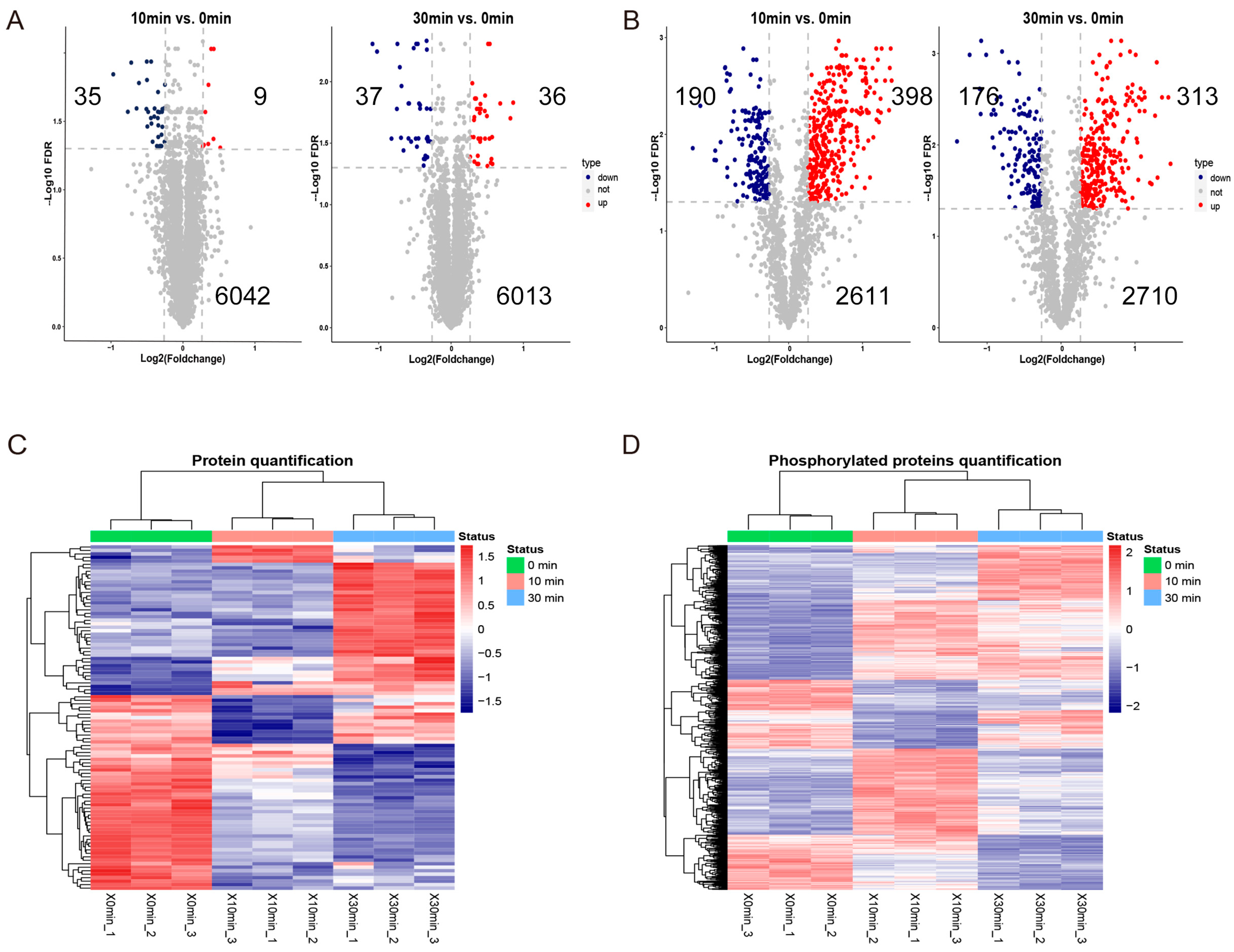
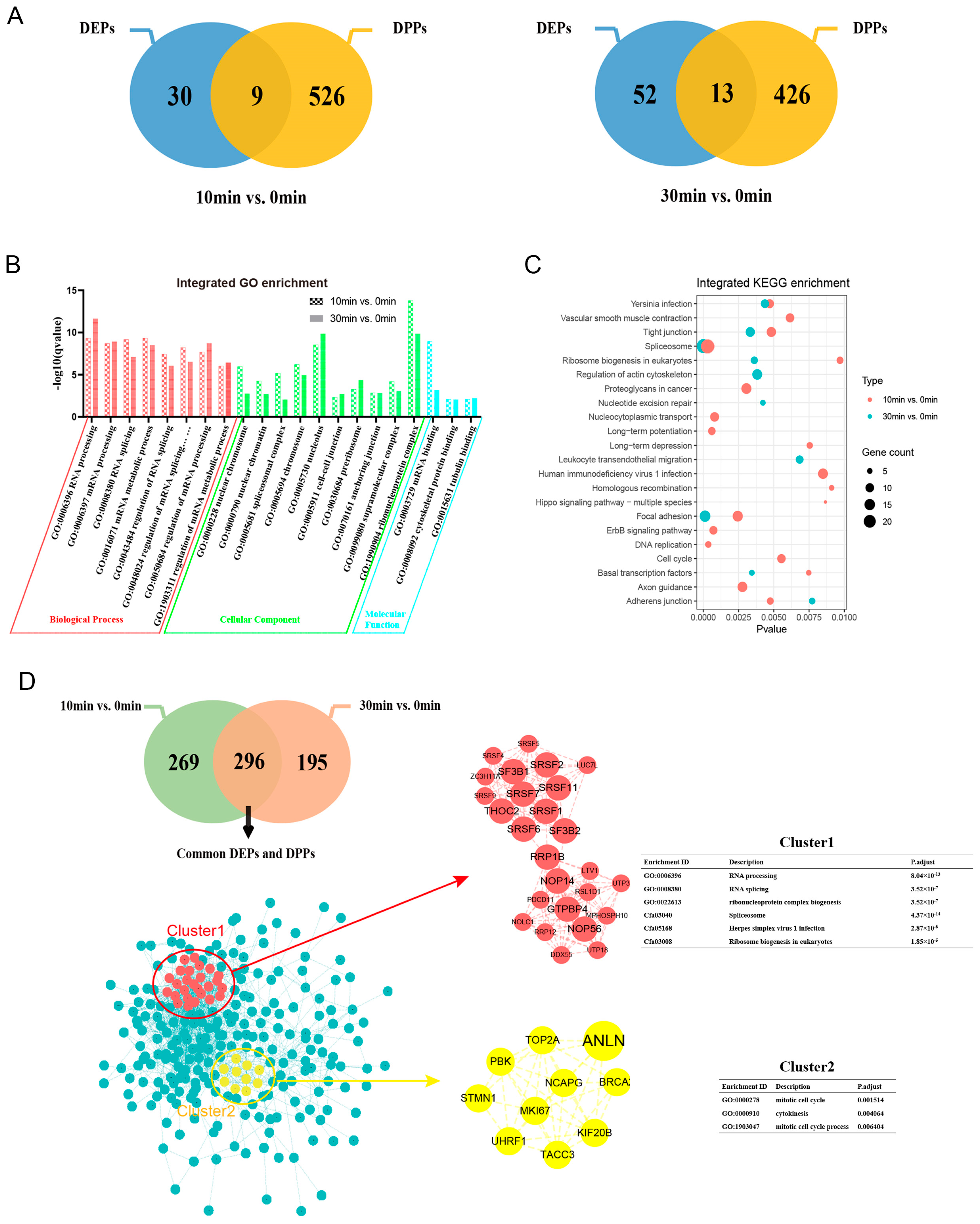
2.2. Screening of Differentially Expressed Proteins and Differentially Phosphorylated Proteins
2.3. Subcellular Localization and KEGG Enrichment Analysis
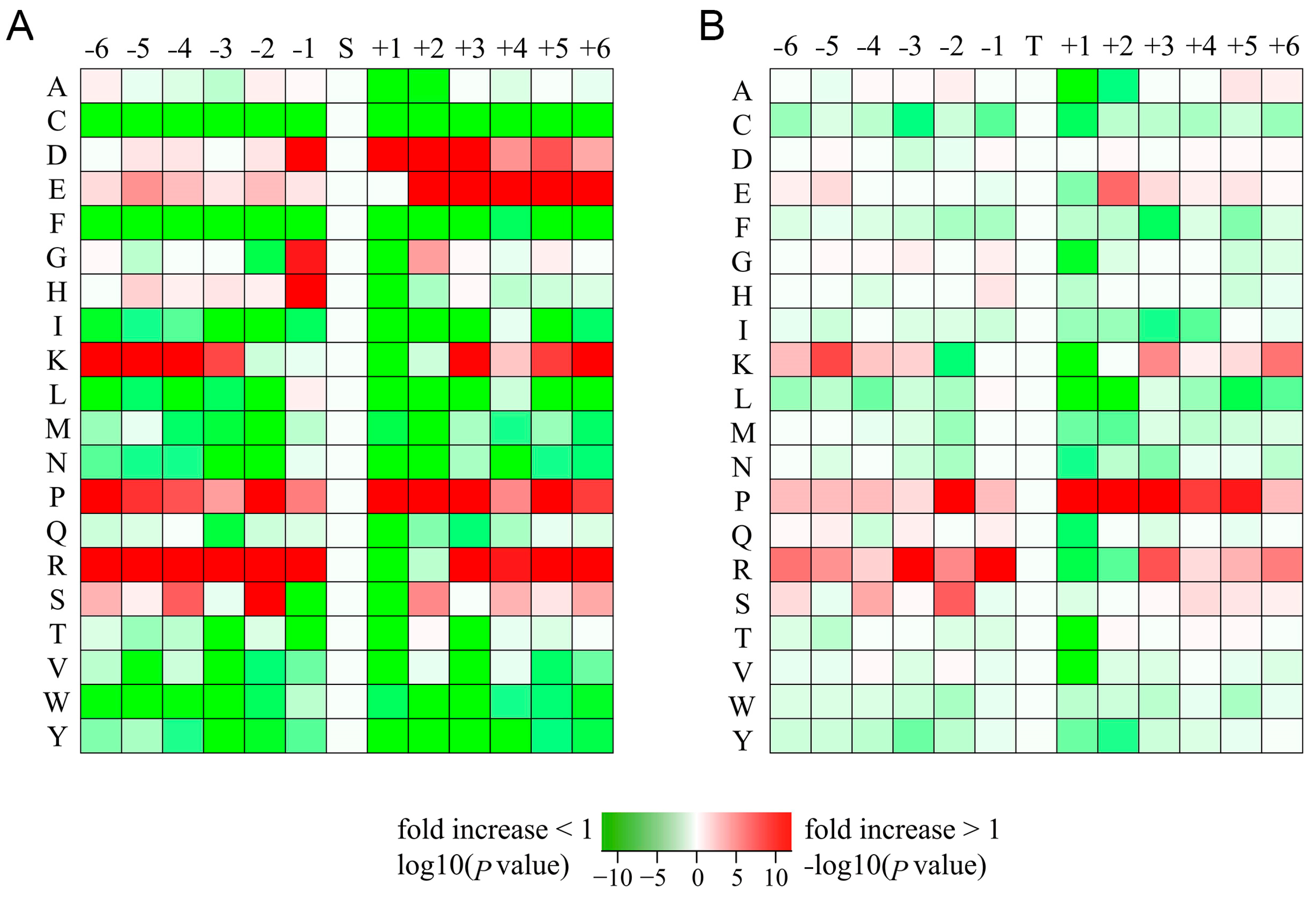
2.4. Motif Analysis of Phosphorylation Sites
2.5. Integration Analysis of Differentially Expressed Proteins and Differentially Phosphorylated Proteins
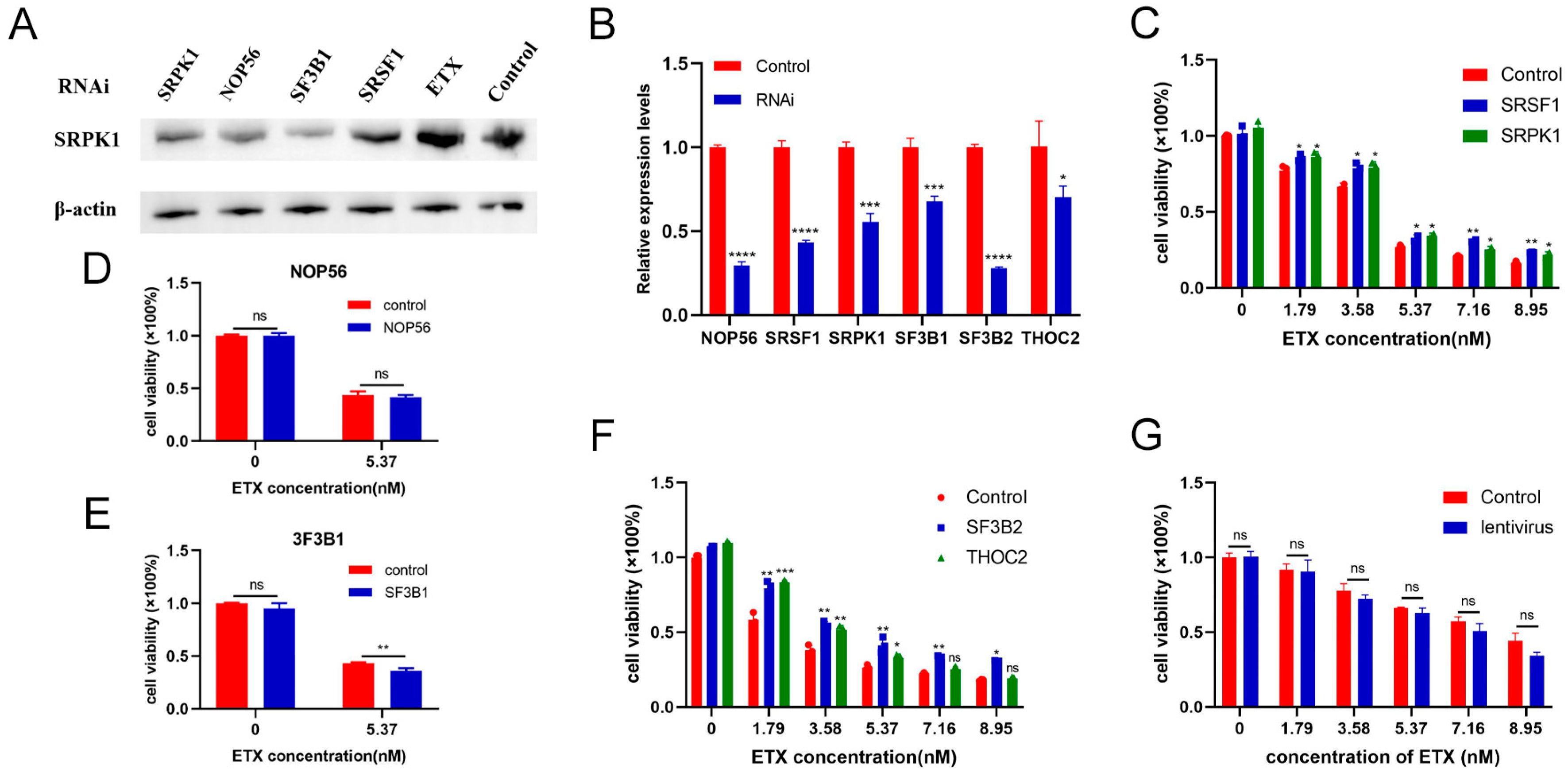
2.6. Inhibitors Reduced Toxicity of ETX on MDCK Cells
2.7. SRSF1 and SRPK1 Inhibit the Toxicity of ETX to MDCK Cells
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Recombinant Toxin and Reagents
5.2. Cell Culture
5.3. Western Blotting
5.4. Protein Extraction
5.5. Trypsin Digestion
5.6. TMT Labeling
5.7. HPLC Fractionation
5.8. LC-MS/MS Analysis
5.9. Database Search
5.10. Bio-Material-Based PTM Enrichment (For Phosphorylation)
5.11. Standardization of Data
5.12. Bioinformatics Analysis
5.13. Cytotoxicity Assay
5.14. Lentivirus-Mediated RNA Interference Assay
5.15. RT-qRCR
5.16. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, J.; Adams, V.; Bannam, T.L.; Miyamoto, K.; Garcia, J.P.; Uzal, F.A.; Rood, J.I.; McClane, B.A. Toxin plasmids of Clostridium perfringens. Microbiol. Mol. Biol. Rev. 2013, 77, 208–233. [Google Scholar] [CrossRef]
- Garcia, J.P.; Giannitti, F.; Finnie, J.W.; Manavis, J.; Beingesser, J.; Adams, V.; Rood, J.I.; Uzal, F.A. Comparative neuropathology of ovine enterotoxemia produced by Clostridium perfringens type D wild-type strain CN1020 and its genetically modified derivatives. Vet. Pathol. 2015, 52, 465–475. [Google Scholar] [CrossRef]
- Cases, M.; Llobet, A.; Terni, B.; de Aranda, I.G.; Blanch, M.; Doohan, B.; Revill, A.; Brown, A.M.; Blasi, J.; Solsona, C. Acute Effect of Pore-Forming Clostridium perfringens ε-Toxin on Compound Action Potentials of Optic Nerve of Mouse. eNeuro 2017, 4, ENEURO.0051-17.2017. [Google Scholar] [CrossRef]
- Yao, W.; Kang, J.; Kang, L.; Gao, S.; Yang, H.; Ji, B.; Li, P.; Liu, J.; Xin, W.; Wang, J. Immunization with a novel Clostridium perfringens epsilon toxin mutant rETX(Y196E)-C confers strong protection in mice. Sci. Rep. 2016, 6, 24162. [Google Scholar] [CrossRef]
- Bokori-Brown, M.; Hall, C.A.; Vance, C.; da Costa, S.P.F.; Savva, C.G.; Naylor, C.E.; Cole, A.R.; Basak, A.K.; Moss, D.S.; Titball, R.W. Clostridium perfringens epsilon toxin mutant Y30A-Y196A as a recombinant vaccine candidate against enterotoxemia. Vaccine 2014, 32, 2682–2687. [Google Scholar] [CrossRef]
- Bokori-Brown, M.; Savva, C.G.; da Costa, S.P.F.; Naylor, C.E.; Basak, A.K.; Titball, R.W. Molecular basis of toxicity of Clostridium perfringens epsilon toxin. FEBS J. 2011, 278, 4589–4601. [Google Scholar] [CrossRef]
- Minami, J.; Katayama, S.; Matsushita, O.; Matsushita, C.; Okabe, A. Lambda-toxin of Clostridium perfringens activates the precursor of epsilon-toxin by releasing its N- and C-terminal peptides. Microbiol. Immunol. 1997, 41, 527–535. [Google Scholar] [CrossRef]
- Stiles, B.G.; Barth, G.; Barth, H.; Popoff, M.R. Clostridium perfringens epsilon toxin: A malevolent molecule for animals and man? Toxins 2013, 5, 2138–2160. [Google Scholar] [CrossRef]
- Popoff, M.R. Epsilon toxin: A fascinating pore-forming toxin. FEBS J. 2011, 278, 4602–4615. [Google Scholar] [CrossRef]
- Petit, L.; Gibert, M.; Gillet, D.; Laurent-Winter, C.; Boquet, P.; Popoff, M.R. Clostridium perfringens epsilon-toxin acts on MDCK cells by forming a large membrane complex. J. Bacteriol. 1997, 179, 6480–6487. [Google Scholar] [CrossRef]
- Shortt, S.J.; Titball, R.W.; Lindsay, C.D. An assessment of the in vitro toxicology of Clostridium perfringens type D epsilon-toxin in human and animal cells. Hum. Exp. Toxicol. 2000, 19, 108–116. [Google Scholar] [CrossRef]
- Ivie, S.E.; Fennessey, C.M.; Sheng, J.; Rubin, D.H.; McClain, M.S. Gene-trap mutagenesis identifies mammalian genes contributing to intoxication by Clostridium perfringens ε-toxin. PLoS ONE 2011, 6, e17787. [Google Scholar] [CrossRef]
- Chassin, C.; Bens, M.; de Barry, J.; Courjaret, R.; Bossu, J.L.; Cluzeaud, F.; Mkaddem, S.B.; Gibert, M.; Poulain, B.; Popoff, M.R.; et al. Pore-forming epsilon toxin causes membrane permeabilization and rapid ATP depletion-mediated cell death in renal collecting duct cells. Am. J. Physiol. Renal Physiol. 2007, 293, F927–F937. [Google Scholar] [CrossRef]
- Gao, J.; Xin, W.; Huang, J.; Ji, B.; Gao, S.; Chen, L.; Kang, L.; Yang, H.; Shen, X.; Zhao, B.; et al. Research articleHemolysis in human erythrocytes by Clostridium perfringens epsilon toxin requires activation of P2 receptors. Virulence 2018, 9, 1601–1614. [Google Scholar] [CrossRef]
- Xin, W.; Wang, J. Clostridium perfringens epsilon toxin: Toxic effects and mechanisms of action. Biosaf. Health 2019, 1, 71–75. [Google Scholar] [CrossRef]
- Savva, C.G.; Clark, A.R.; Naylor, C.E.; Popoff, M.R.; Moss, D.S.; Basak, A.K.; Titball, R.W.; Bokori-Brown, M. The pore structure of Clostridium perfringens epsilon toxin. Nat. Commun. 2019, 10, 2641. [Google Scholar] [CrossRef]
- Miyata, S.; Minami, J.; Tamai, E.; Matsushita, O.; Shimamoto, S.; Okabe, A. Clostridium perfringens epsilon-toxin forms a heptameric pore within the detergent-insoluble microdomains of Madin-Darby canine kidney cells and rat synaptosomes. J. Biol. Chem. 2002, 277, 39463–39468. [Google Scholar] [CrossRef]
- Petit, L.; Maier, E.; Gibert, M.; Popoff, M.R.; Benz, R. Clostridium perfringens epsilon toxin induces a rapid change of cell membrane permeability to ions and forms channels in artificial lipid bilayers. J. Biol. Chem. 2001, 276, 15736–15740. [Google Scholar] [CrossRef]
- Ferrarezi, M.C.; Curci, V.C.; Cardoso, T.C. Cellular vacuolation and mitochondrial-associated factors induced by Clostridium perfringens epsilon toxin detected using acoustic flow cytometry. Anaerobe 2013, 24, 55–59. [Google Scholar] [CrossRef]
- Wioland, L.; Dupont, J.L.; Doussau, F.; Gaillard, S.; Heid, F.; Isope, P.; Pauillac, S.; Popoff, M.R.; Bossu, J.L.; Poulain, B. Epsilon toxin from Clostridium perfringens acts on oligodendrocytes without forming pores, and causes demyelination. Cell Microbiol. 2015, 17, 369–388. [Google Scholar] [CrossRef]
- Rashid Rumah, K.; Eleso, O.; Fischetti, V.A.J.B. Human blood exposure to Clostridium perfringens epsilon toxin may shed light on erythrocyte fragility during active multiple sclerosis. bioRivx 2019, 789123. [Google Scholar] [CrossRef]
- Abou Faycal, C.; Gazzeri, S.; Eymin, B. A VEGF-A/SOX2/SRSF2 network controls VEGFR1 pre-mRNA alternative splicing in lung carcinoma cells. Sci. Rep. 2019, 9, 336. [Google Scholar] [CrossRef]
- He, C.; Liu, B.; Wang, H.Y.; Wu, L.; Zhao, G.; Huang, C.; Liu, Y.; Shan, B.; Liu, L. Inhibition of SRPK1, a key splicing regulator, exhibits antitumor and chemotherapeutic-sensitizing effects on extranodal NK/T-cell lymphoma cells. BMC Cancer 2022, 22, 1100. [Google Scholar] [CrossRef]
- Freedman, J.C.; McClane, B.A.; Uzal, F.A. New insights into Clostridium perfringens epsilon toxin activation and action on the brain during enterotoxemia. Anaerobe 2016, 41, 27–31. [Google Scholar] [CrossRef]
- Babele, P.; Verma, S.; Kumar, R.B.; Bhagyawant, S.S.; Kamboj, D.V.; Alam, S.I. Elucidation of protein biomarkers in plasma and urine for epsilon toxin exposure in mouse model. Anaerobe 2019, 59, 76–91. [Google Scholar] [CrossRef]
- Berget, S.M.; Moore, C.; Sharp, P.A. Spliced segments at the 5′ terminus of adenovirus 2 late mRNA. Proc. Natl. Acad. Sci. USA 1977, 74, 3171–3175. [Google Scholar] [CrossRef]
- Chow, L.T.; Gelinas, R.E.; Broker, T.R.; Roberts, R.J. An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell 1977, 12, 1–8. [Google Scholar] [CrossRef]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes. Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef]
- Ma, X.; He, F. Advances in the study of SR protein family. Genom. Proteom. Bioinform. 2003, 1, 2–8. [Google Scholar] [CrossRef]
- Lv, Y.; Zhang, W.; Zhao, J.; Sun, B.; Qi, Y.; Ji, H.; Chen, C.; Zhang, J.; Sheng, J.; Wang, T.; et al. SRSF1 inhibits autophagy through regulating Bcl-x splicing and interacting with PIK3C3 in lung cancer. Signal Transduct. Target. Ther. 2021, 6, 108. [Google Scholar] [CrossRef]
- Du, J.X.; Luo, Y.H.; Zhang, S.J.; Wang, B.; Chen, C.; Zhu, G.Q.; Zhu, P.; Cai, C.Z.; Wan, J.L.; Cai, J.L.; et al. Splicing factor SRSF1 promotes breast cancer progression via oncogenic splice switching of PTPMT1. J. Exp. Clin. Cancer Res. 2021, 40, 171. [Google Scholar] [CrossRef]
- Wan, L.; Lin, K.T.; Rahman, M.A.; Ishigami, Y.; Wang, Z.; Jensen, M.A.; Wilkinson, J.E.; Park, Y.; Tuveson, D.A.; Krainer, A.R. Splicing Factor SRSF1 Promotes Pancreatitis and KRASG12D-Mediated Pancreatic Cancer. Cancer Discov. 2023, 13, 1678–1695. [Google Scholar] [CrossRef]
- Lance, A.; Druhan, L.J.; Vestal, C.G.; Steuerwald, N.M.; Hamilton, A.; Smith, M.; Price, A.; Tjaden, E.; Fox, A.N.; Avalos, B.R. Altered expression of CSF3R splice variants impacts signal response and is associated with SRSF2 mutations. Leukemia 2020, 34, 369–379. [Google Scholar] [CrossRef]
- Gout, S.; Brambilla, E.; Boudria, A.; Drissi, R.; Lantuejoul, S.; Gazzeri, S.; Eymin, B. Abnormal expression of the pre-mRNA splicing regulators SRSF1, SRSF2, SRPK1 and SRPK2 in non small cell lung carcinoma. PLoS ONE 2012, 7, e46539. [Google Scholar] [CrossRef]
- Luo, C.; Cheng, Y.; Liu, Y.; Chen, L.; Liu, L.; Wei, N.; Xie, Z.; Wu, W.; Feng, Y. SRSF2 Regulates Alternative Splicing to Drive Hepatocellular Carcinoma Development. Cancer Res. 2017, 77, 1168–1178. [Google Scholar] [CrossRef]
- Li, K.; Wang, Z. Splicing factor SRSF2-centric gene regulation. Int. J. Biol. Sci. 2021, 17, 1708–1715. [Google Scholar] [CrossRef]
- Wan, L.; Yu, W.; Shen, E.; Sun, W.; Liu, Y.; Kong, J.; Wu, Y.; Han, F.; Zhang, L.; Yu, T.; et al. SRSF6-regulated alternative splicing that promotes tumour progression offers a therapy target for colorectal cancer. Gut 2019, 68, 118–129. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, H.; Yu, J.; Yao, X.; Yang, S.; Li, W.; Xu, L.; Zhao, L. LncRNA CRNDE attenuates chemoresistance in gastric cancer via SRSF6-regulated alternative splicing of PICALM. Mol. Cancer 2021, 20, 6. [Google Scholar] [CrossRef]
- Li, Y.; Xu, J.; Lu, Y.; Bian, H.; Yang, L.; Wu, H.; Zhang, X.; Zhang, B.; Xiong, M.; Chang, Y.; et al. DRAK2 aggravates nonalcoholic fatty liver disease progression through SRSF6-associated RNA alternative splicing. Cell Metab. 2021, 33, 2004–2020.e9. [Google Scholar] [CrossRef]
- Makishima, H.; Visconte, V.; Sakaguchi, H.; Jankowska, A.M.; Kar, S.A.; Jerez, A.; Przychodzen, B.; Bupathi, M.; Guinta, K.; Afable, M.G.; et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 2012, 119, 3203–3210. [Google Scholar] [CrossRef]
- Cazzola, M.; Rossi, M.; Malcovati, L. Biologic and clinical significance of somatic mutations of SF3B1 in myeloid and lymphoid neoplasms. Blood 2013, 121, 260–269. [Google Scholar] [CrossRef]
- Seo, J.Y.; Lee, K.O.; Kim, S.H.; Kim, K.; Jung, C.W.; Jang, J.H.; Kim, H.J. Clinical significance of SF3B1 mutations in Korean patients with myelodysplastic syndromes and myelodysplasia/myeloproliferative neoplasms with ring sideroblasts. Ann. Hematol. 2014, 93, 603–608. [Google Scholar] [CrossRef]
- Bullock, N.; Oltean, S. The many faces of SRPK1. J. Pathol. 2017, 241, 437–440. [Google Scholar] [CrossRef]
- Liu, H.; Gong, Z.; Li, K.; Zhang, Q.; Xu, Z.; Xu, Y. SRPK1/2 and PP1α exert opposite functions by modulating SRSF1-guided MKNK2 alternative splicing in colon adenocarcinoma. J. Exp. Clin. Cancer Res. 2021, 40, 75. [Google Scholar] [CrossRef]
- Patel, M.; Sachidanandan, M.; Adnan, M. Serine arginine protein kinase 1 (SRPK1): A moonlighting protein with theranostic ability in cancer prevention. Mol. Biol. Rep. 2019, 46, 1487–1497. [Google Scholar] [CrossRef]
- Geng, Z.; Huang, J.; Kang, L.; Gao, S.; Yuan, Y.; Li, Y.; Wang, J.; Xin, W.; Wang, J. Clostridium perfringens epsilon toxin binds to erythrocyte MAL receptors and triggers phosphatidylserine exposure. J. Cell Mol. Med. 2020, 24, 7341–7352. [Google Scholar] [CrossRef]
- Zhang, N.; Jiang, N.; Zhang, K.; Zheng, L.; Zhang, D.; Sang, X.; Feng, Y.; Chen, R.; Yang, N.; Wang, X.; et al. Landscapes of Protein Posttranslational Modifications of African Trypanosoma Parasites. iScience 2020, 23, 101074. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Accession | Protein Description | Gene me | FC10 | FC30 | FDR10 | FDR30 | Type |
|---|---|---|---|---|---|---|---|
| E2RED7 | Bromodomain adjacent to zinc finger domain 1B | BAZ1B | 1.328 | 1.33 | 0.009233 | 0.01666 | up |
| J9PAY8 | DNA topoisomerase 2 | TOP2A | 1.295 | 1.257 | 0.009233 | 0.013718 | up |
| E2R5T9 | Actinin alpha 4 | ACTN4 | 0.727 | 0.665 | 0.011489 | 0.015039 | down |
| F1PLS4 | Vimentin | VIM | 0.697 | 0.595 | 0.011489 | 0.016655 | down |
| E2RNB0 | Histone H2A | H2AX | 0.505 | 0.471 | 0.014266 | 0.004933 | down |
| L7N0L3 | Histone H4 | H4C7 | 0.699 | 0.598 | 0.015655 | 0.004933 | down |
| E2RR73 | Histone cluster 1 H1 family member a | HIST1H1A | 0.647 | 0.492 | 0.016482 | 0.005695 | down |
| E2QY07 | Actinin alpha 1 | ACTN1 | 0.831 | 0.727 | 0.016911 | 0.004933 | down |
| F1Q0N9 | Keratin 19 | KRT19 | 0.753 | 0.699 | 0.025294 | 0.005341 | down |
| F1Q272 | WAS/WASL interacting protein family member 2 | WIPF2 | 0.633 | 0.665 | 0.025294 | 0.028942 | down |
| F1PB61 | TATA-box binding protein associated factor 15 | TAF15 | 0.812 | 0.713 | 0.025522 | 0.029035 | down |
| F1PRB0 | IF rod domain- containing protein | KRT86 | 0.696 | 0.622 | 0.025522 | 0.010853 | down |
| F1PW86 | Inositol 1,4,5-trisphosphate receptor type 1 | ITPR1 | 0.778 | 0.721 | 0.026721 | 0.033783 | down |
| E2REU6 | F rod domain- containing protein | KRT18 | 0.751 | 0.669 | 0.028972 | 0.015039 | down |
| E2RJ54 | Heterogeneous nuclear ribonucleoprotein K | HNRNPK | 0.727 | 0.792 | 0.029708 | 0.029035 | down |
| L7N071 | Actinin alpha 4 | ACTN4 | 0.777 | 0.732 | 0.029708 | 0.028942 | down |
| E2R6 | PEST proteolytic sigl containing nuclear protein | PCNP | 0.703 | 0.612 | 0.034266 | 0.007628 | down |
| Protein Accession | Protein Description | Gene me | Phosphosites | FC10 | FC30 | FDR10 | FDR30 | Type |
|---|---|---|---|---|---|---|---|---|
| F1PLN6 | Tripartite motif containing 33 | TRIM33 | S987 | 0.181 | 0.104 | 0.006239 | 0.03292 | down |
| F1PU14 | PDGFA associated protein 1 | PDAP1 | Y71 | 0.202 | 0.252 | 0.00203 | 0.002436 | down |
| F6XZH6 | Ladinin 1 | LAD1 | S462 | 0.503 | 0.271 | 0.014744 | 0.001249 | down |
| F1Q2Z4 | Serine/arginine repetitive matrix 2 | SRRM2 | S2394 | 0.728 | 0.327 | 0.017377 | 0.003296 | down |
| F1PKT2 | Lipolysis stimulated lipoprotein receptor | LSR | S592 | 0.437 | 0.333 | 0.005074 | 0.001034 | down |
| J9P969 | AHK nucleoprotein | AHK | S4691 | 3.607 | 3.563 | 0.003419 | 0.007272 | up |
| E2RR55 | Nuclear factor of activated T cells 2 interacting protein | NFATC2IP | S90 | 2.512 | 3.230 | 0.03395 | 0.000727 | up |
| J9NRJ1 | 40S ribosomal protein S6 | --- | S235 | 2.604 | 3.169 | 0.001298 | 0.001109 | up |
| E2RJ15 | Myotubularin related protein 10 | MTMR10 | S600 | 4.211 | 3.095 | 0.001298 | 0.003654 | up |
| J9NRJ1 | 40S ribosomal protein S6 | --- | S236 | 2.278 | 3.077 | 0.00203 | 0.000994 | up |
| Gene me | Protein Accession | Type10 | Type30 | Subcellular Localization | KEGG Pathway |
|---|---|---|---|---|---|
| SRSF1 | E2RJL3 | down | down | nucleus | cfa04657; cfa05168; cfa03040 |
| SRSF2 | E2RDB2 | up | up | nucleus | cfa05168; cfa03040 |
| SRSF6 | F1PTE0 | up | up | nucleus | cfa05168; cfa03040 |
| SRSF7 | J9P2I8 | down | up | nucleus | cfa05168; cfa03040 |
| SF3B1 | F1PKF6 | up | up | plasma membrane | cfa03040 |
| SF3B2 | E2RL65 | up | up | nucleus | cfa03040 |
| GTPBP4 | F1PJY9 | up | up | nucleus | cfa03008 |
| THOC2 | J9PB31 | down | down | cytoplasm | cfa03013; cfa03040 |
| NOP56 | E2QU53 | up | up | nucleus | cfa03008 |
| NOP14 | F1PI93 | up | up | plasma membrane | 0 |
| ANLN | F1PZ90 | up | up | nucleus | 0 |
| SRSF11 | F1P6U8 | down | down | nucleus | 0 |
| RRP1B | F1PS87 | down | down | cytoplasm, nucleus | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yue, N.; Huang, J.; Dong, M.; Li, J.; Gao, S.; Wang, J.; Wang, Y.; Li, D.; Luo, X.; Liu, T.; et al. Proteome and Phosphoproteome Profiling Reveal the Toxic Mechanism of Clostridium perfringens Epsilon Toxin in MDCK Cells. Toxins 2024, 16, 394. https://doi.org/10.3390/toxins16090394
Yue N, Huang J, Dong M, Li J, Gao S, Wang J, Wang Y, Li D, Luo X, Liu T, et al. Proteome and Phosphoproteome Profiling Reveal the Toxic Mechanism of Clostridium perfringens Epsilon Toxin in MDCK Cells. Toxins. 2024; 16(9):394. https://doi.org/10.3390/toxins16090394
Chicago/Turabian StyleYue, Nan, Jing Huang, Mingxin Dong, Jiaxin Li, Shan Gao, Jing Wang, Yingshuang Wang, Dongxue Li, Xi Luo, Tingting Liu, and et al. 2024. "Proteome and Phosphoproteome Profiling Reveal the Toxic Mechanism of Clostridium perfringens Epsilon Toxin in MDCK Cells" Toxins 16, no. 9: 394. https://doi.org/10.3390/toxins16090394
APA StyleYue, N., Huang, J., Dong, M., Li, J., Gao, S., Wang, J., Wang, Y., Li, D., Luo, X., Liu, T., Han, S., Dong, L., Chen, M., Wang, J., Xu, N., Kang, L., & Xin, W. (2024). Proteome and Phosphoproteome Profiling Reveal the Toxic Mechanism of Clostridium perfringens Epsilon Toxin in MDCK Cells. Toxins, 16(9), 394. https://doi.org/10.3390/toxins16090394