β-N-Methylamino-L-Alanine Induces Neurological Deficits and Shortened Life Span in Drosophila

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Drosophila Culture and Drug Feeding
2.2. Negative Geotaxic Assay
2.3. Aversive Phototaxic Suppression Assay
2.4. Tissue Sample Preparation for HPLC
2.5. Detection of BMAA with HPLC Analysis
3. Results
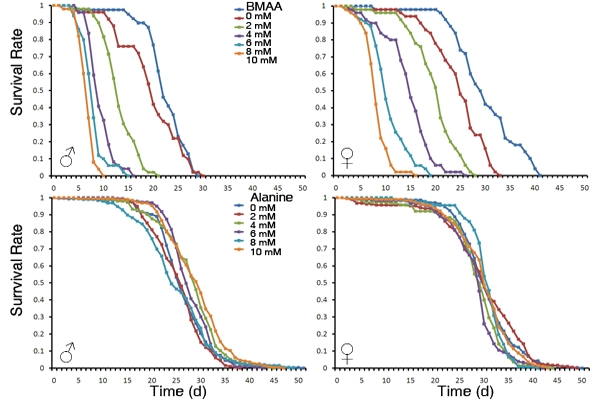
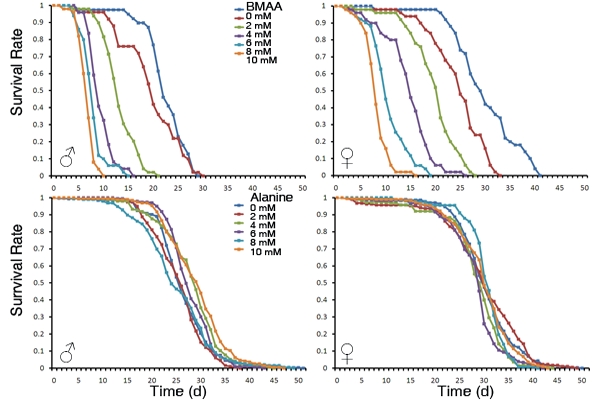
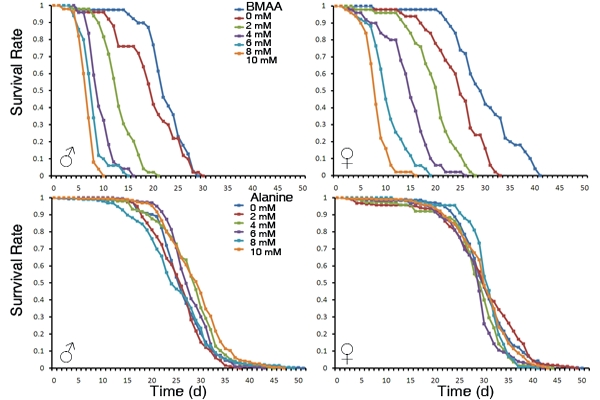
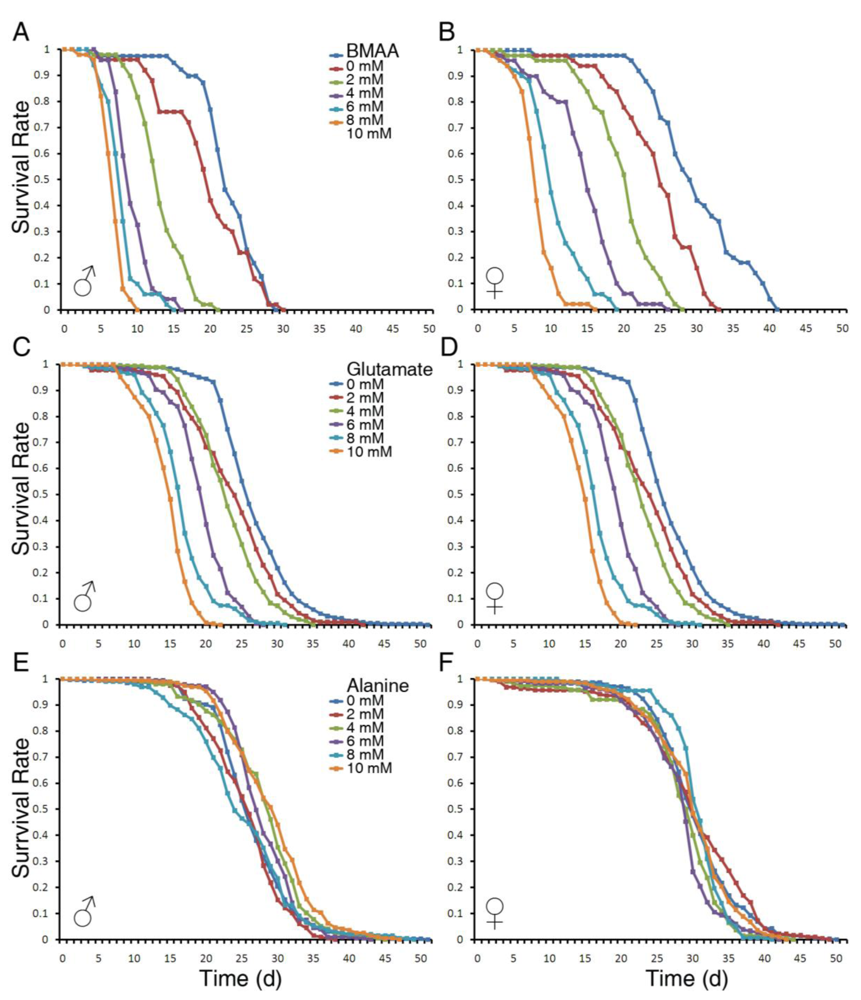
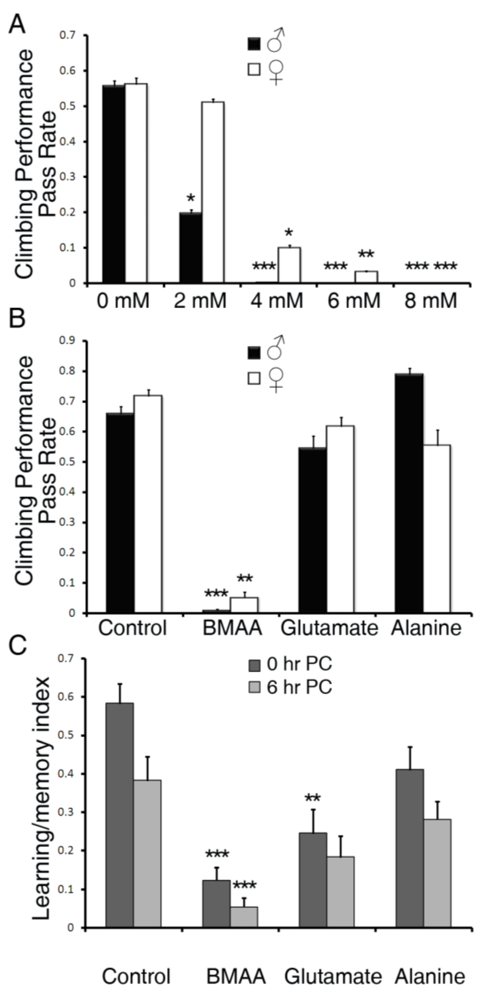
3.1. Dietary Intake of BMAA Reduced Life Span and Induced Locomotor and Learning and Memory Deficits in a Dose-dependent Manner


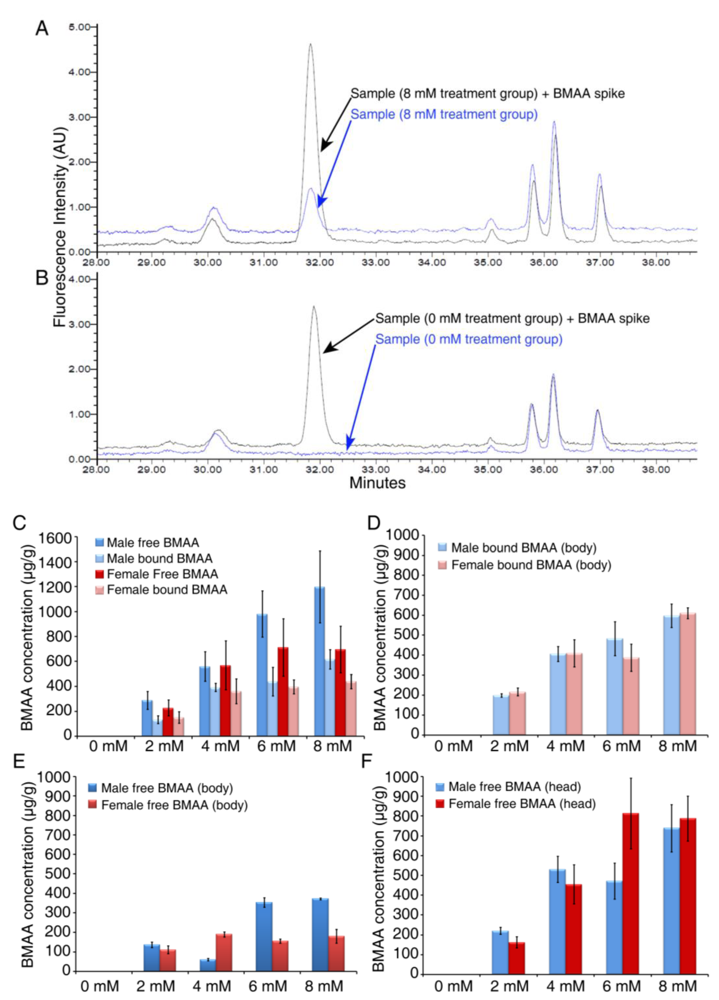
3.2. Dose-dependent Accumulation of BMAA in Flies

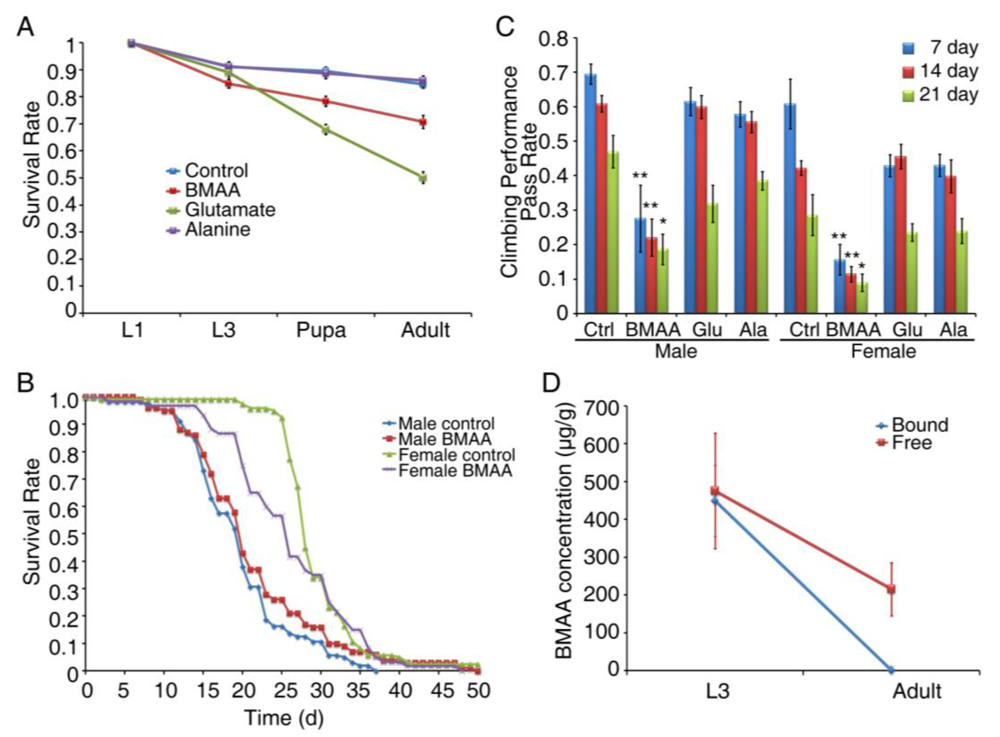
3.3. Developmental Exposure to BMAA Induces Delayed Locomotor Deficits in Aged Flies

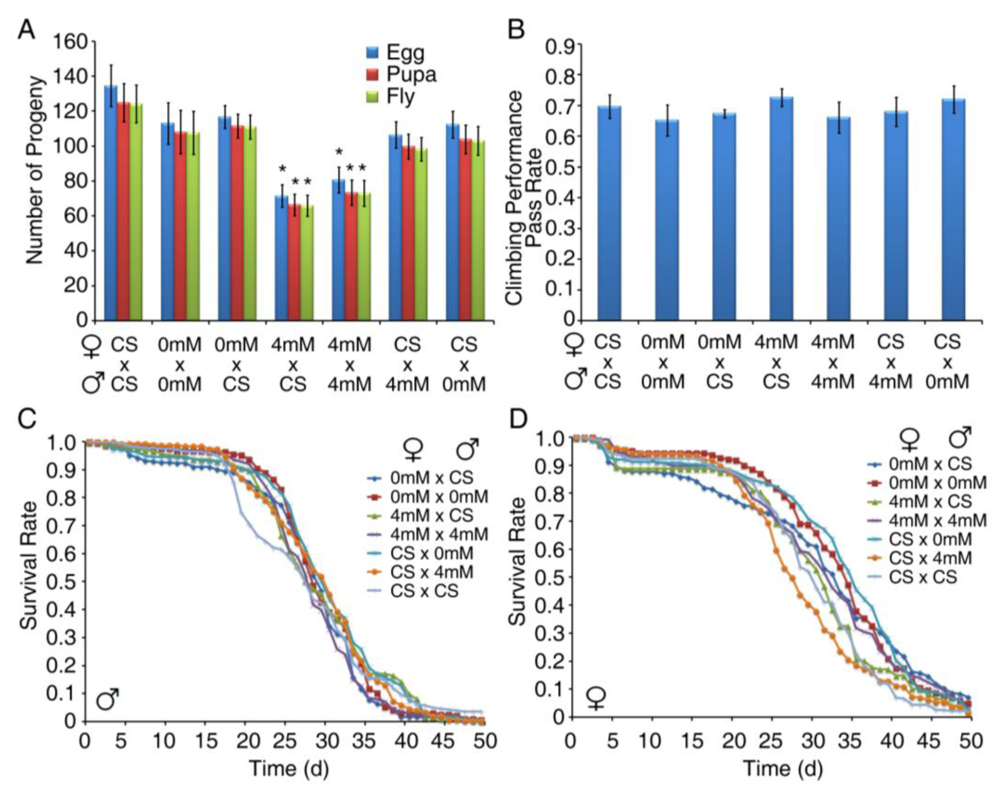
3.4. Developmental Exposure to BMAA Reduces Fertility in Female Flies

4. Discussions
4.1. Toxic Levels of BMAA in Brain Tissue
4.2. Progressive Toxicity of BMAA
4.3. BMAA Has Stronger Toxicity than Glutamate
5. Conclusions
Acknowledgements
References and Notes
- Cox, P.A.; Sacks, O.W. Cycad neurotoxins, consumption of flying foxes, and ALS-PDC disease in Guam. Neurology 2002, 58, 956–959. [Google Scholar] [PubMed]
- Kisby, G.E.; Ellison, M.; Spencer, P.S. Content of the neurotoxins cycasin (methylazoxymethanol beta-D-glucoside) and BMAA (beta-N-methylamino-L-alanine) in cycad flour prepared by Guam Chamorros. Neurology 1992, 42, 1336–1340. [Google Scholar]
- Cox, P.A.; Banack, S.A.; Murch, S.J. Biomagnification of cyanobacterial neurotoxins and neurodegenerative disease among the Chamorro people of Guam. Proc. Natl. Acad. Sci. USA 2003, 100, 13380–13383. [Google Scholar]
- Murch, S.J.; Cox, P.A.; Banack, S.A. A mechanism for slow release of biomagnified cyanobacterial neurotoxins and neurodegenerative disease in Guam. Proc. Natl. Acad. Sci. USA 2004, 101, 12228–12231. [Google Scholar]
- Banack, S.A.; Johnson, H.E.; Cheng, R.; Cox, P.A. Production of the Neurotoxin BMAA by a Marine Cyanobacterium. Mar. Drugs 2007, 5, 180–196. [Google Scholar]
- Metcalf, J.S.; Banack, S.A.; Lindsay, J.; Morrison, L.F.; Cox, P.A.; Codd, G.A. Co-occurrence of beta-N-methylamino-L-alanine, a neurotoxic amino acid with other cyanobacterial toxins in British waterbodies, 1990–2004. Environ. Microbiol. 2008, 10, 702–708. [Google Scholar]
- Jonasson, S.; Eriksson, J.; Berntzon, L.; Spacil, Z.; Ilag, L.L.; Ronnevi, L.O.; Rasmussen, U.; Bergman, B. Transfer of a cyanobacterial neurotoxin within a temperate aquatic ecosystem suggests pathways for human exposure. Proc. Natl. Acad. Sci. USA 2010, 107, 9252–9257. [Google Scholar]
- Johnson, H.E.; King, S.R.; Banack, S.A.; Webster, C.; Callanaupa, W.J.; Cox, P.A. Cyanobacteria (Nostoc commune) used as a dietary item in the Peruvian highlands produce the neurotoxic amino acid BMAA. J. Ethnopharmacol. 2008, 118, 159–165. [Google Scholar]
- Mash, D.C. Cyanobacterial toxins in neurodegeneration. Continuum Lifelong Learn. Neurol. 2008, 14, 138–149. [Google Scholar]
- Murch, S.J.; Cox, P.A.; Banack, S.A.; Steele, J.C.; Sacks, O.W. Occurrence of beta-methylamino-l-alanine (BMAA) in ALS/PDC patients from Guam. Acta Neurol. Scand. 2004, 110, 267–269. [Google Scholar]
- Pablo, J.; Banack, S.A.; Cox, P.A.; Johnson, T.E.; Papapetropoulos, S.; Bradley, W.G.; Buck, A.; Mash, D.C. Cyanobacterial neurotoxin BMAA in ALS and Alzheimer's disease. Acta Neurol. Scand. 2009. [Google Scholar]
- Papapetropoulos, S. Is there a role for naturally occurring cyanobacterial toxins in neurodegeneration? The beta-N-methylamino-L-alanine (BMAA) paradigm. Neurochem. Int. 2007, 50, 998–1003. [Google Scholar]
- Snyder, L.R.; Cruz-Aguado, R.; Sadilek, M.; Galasko, D.; Shaw, C.A.; Montine, T.J. Lack of cerebral bmaa in human cerebral cortex. Neurology 2009, 72, 1360–1361. [Google Scholar]
- Lobner, D.; Piana, P.M.; Salous, A.K.; Peoples, R.W. Beta-N-methylamino-L-alanine enhances neurotoxicity through multiple mechanisms. Neurobiol. Dis. 2007, 25, 360–366. [Google Scholar]
- Rao, S.D.; Banack, S.A.; Cox, P.A.; Weiss, J.H. BMAA selectively injures motor neurons via AMPA/kainate receptor activation. Exp. Neurol. 2006, 201, 244–252. [Google Scholar]
- Weiss, J.H.; Christine, C.W.; Choi, D.W. Bicarbonate dependence of glutamate receptor activation by beta-N-methylamino-L-alanine: Channel recording and study with related compounds. Neuron 1989, 3, 321–326. [Google Scholar]
- Brownson, D.M.; Mabry, T.J.; Leslie, S.W. The cycad neurotoxic amino acid, beta-N-methylamino-L-alanine (BMAA), elevates intracellular calcium levels in dissociated rat brain cells. J. Ethnopharmacol. 2002, 82, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Aguado, R.; Winkler, D.; Shaw, C.A. Lack of behavioral and neuropathological effects of dietary beta-methylamino-L-alanine (BMAA) in mice. Pharmacol. Biochem. Behav. 2006, 84, 294–299. [Google Scholar]
- Karamyan, V.T.; Speth, R.C. Animal models of BMAA neurotoxicity: A critical review. Life Sci. 2008, 82, 233–246. [Google Scholar]
- Zhou, X.; Escala, W.; Papapetropoulos, S.; Bradley, W.G.; Zhai, R.G. BMAA neurotoxicity in Drosophila. Amyotroph. Lateral. Scler. 2009, 10, 61–66. [Google Scholar]
- Ganetzky, B.; Flanagan, J.R. On the relationship between senescence and age-related changes in two wild-type strains of Drosophila melanogaster. Exp. Gerontol. 1978, 13, 189–196. [Google Scholar]
- Le Bourg, E.; Lints, F.A. Hypergravity and aging in Drosophila melanogaster. 4. Climbing activity. Gerontology 1992, 38, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Le Bourg, E.; Buecher, C. Learned suppression of photopositive tendencies in Drosophila melanogaster. Anim. Learn. Behav. 2002, 30, 330–341. [Google Scholar]
- Seugnet, L.; Suzuki, Y.; Stidd, R.; Shaw, P.J. Aversive phototaxic suppression: Evaluation of a short-term memory assay in Drosophila melanogaster. Genes Brain Behav. 2009, 8, 377–389. [Google Scholar]
- Sang, T.K.; Jackson, G.R. Drosophila models of neurodegenerative disease. NeuroRx 2005, 2, 438–446. [Google Scholar]
- Ratnaparkhi, A.; Lawless, G.M.; Schweizer, F.E.; Golshani, P.; Jackson, G.R. A Drosophila model of ALS: Human ALS-associated mutation in VAP33A suggests a dominant negative mechanism. PLoS ONE 2008, 3, e2334. [Google Scholar]
- Tsuda, H.; Han, S.M.; Yang, Y.; Tong, C.; Lin, Y.Q.; Mohan, K.; Haueter, C.; Zoghbi, A.; Harati, Y.; Kwan, J.; Miller, M.A.; Bellen, H.J. The amyotrophic lateral sclerosis 8 protein VAPB is cleaved, secreted, and acts as a ligand for Eph receptors. Cell 2008, 133, 963–977. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.R.; Lagow, R.D.; Xu, K.; Zhang, B.; Bonini, N.M. A drosophila model for amyotrophic lateral sclerosis reveals motor neuron damage by human SOD1. J. Biol. Chem. 2008, 283, 24972–24981. [Google Scholar]
- Benzer, S. From the gene to behavior. Jama 1971, 218, 1015–1022. [Google Scholar]
- Benzer, S. Genetic dissection of behavior. Sci. Am. 1973, 229, 24–37. [Google Scholar]
- Hotta, Y.; Benzer, S. Mapping of behavior in Drosophila mosaics. Symp. Soc. Dev. Biol. 1973, 31, 129–167. [Google Scholar]
- Hirsch, J.; Boudreau, J.C. Studies in experimental behavior genetics. I. The heritability of phototaxis in a population of Drosophila melanogaster. J. Comp. Physiol. Psychol. 1958, 51, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Hendel, T.; Michels, B.; Neuser, K.; Schipanski, A.; Kaun, K.; Sokolowski, M.B.; Marohn, F.; Michel, R.; Heisenberg, M.; Gerber, B. The carrot, not the stick: appetitive rather than aversive gustatory stimuli support associative olfactory learning in individually assayed Drosophila larvae. J. Comp. Physiol. A Neuroethol. Sens. Neural. Behav. Physiol. 2005, 191, 265–279. [Google Scholar]
- Le Bourg, E. Effects of aging on learned suppression of photopositive tendencies in Drosophila melanogaster. Neurobiol. Aging 2004, 25, 1241–1252. [Google Scholar]
- Meunier, N.; Marion-Poll, F.; Rospars, J.P.; Tanimura, T. Peripheral coding of bitter taste in Drosophila. J. Neurobiol. 2003, 56, 139–152. [Google Scholar]
- Quinn, W.G.; Harris, W.A.; Benzer, S. Conditioned behavior in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 1974, 71, 708–712. [Google Scholar]
- Bradley, W.G.; Mash, D.C. Beyond Guam: The cyanobacteria/BMAA hypothesis of the cause of ALS and other neurodegenerative diseases. Amyotroph. Lateral. Scler. 2009, 10, 7–20. [Google Scholar]
- Weiss, J.H.; Koh, J.Y.; Choi, D.W. Neurotoxicity of beta-N-methylamino-L-alanine (BMAA) and beta-N-oxalylamino-L-alanine (BOAA) on cultured cortical neurons. Brain Res. 1989, 497, 64–71. [Google Scholar]
- Ross, S.M.; Seelig, M.; Spencer, P.S. Specific antagonism of excitotoxic action of 'uncommon' amino acids assayed in organotypic mouse cortical cultures. Brain Res. 1987, 425, 120–127. [Google Scholar]
- Buenz, E.J.; Howe, C.L. Beta-methylamino-alanine (BMAA) injures hippocampal neurons in vivo. Neurotoxicology 2007, 28, 702–704. [Google Scholar]
- Cha, J.H.; Makowiec, R.L.; Penney, J.B.; Young, A.B. L-[3H] glutamate labels the metabotropic excitatory amino acid receptor in rodent brain. Neurosci. Lett. 1990, 113, 78–83. [Google Scholar]
- Copani, A.; Canonico, P.L.; Catania, M.V.; Aronica, E.; Bruno, V.; Ratti, E.; van Amsterdam, F.T.; Gaviraghi, G.; Nicoletti, F. Interaction between beta-N-methylamino-L-alanine and excitatory amino acid receptors in brain slices and neuronal cultures. Brain Res. 1991, 558, 79–86. [Google Scholar]
- Rakonczay, Z.; Matsuoka, Y.; Giacobini, E. Effects of L-beta-N-methylamino-L-alanine (L-BMAA) on the cortical cholinergic and glutamatergic systems of the rat. J. Neurosci. Res. 1991, 29, 121–126. [Google Scholar]
- Liu, X.; Rush, T.; Zapata, J.; Lobner, D. beta-N-methylamino-l-alanine induces oxidative stress and glutamate release through action on system Xc(-). Exp. Neurol. 2009, 217, 429–433. [Google Scholar]
- Lobner, D. Mechanisms of beta-N-methylamino-L-alanine induced neurotoxicity. Amyotroph. Lateral. Scler. 2009, 10, 56–60. [Google Scholar]
- Nunn, P.B. Three phases of research on beta-N-methylamino-L-alanine (BMAA)—A neurotoxic amino acid. Amyotroph. Lateral. Scler. 2009, 10, 26–33. [Google Scholar]
- Nunn, P.B.; Ponnusamy, M. Beta-N-methylaminoalanine (BMAA): Metabolism and metabolic effects in model systems and in neural and other tissues of the rat in vitro. Toxicon 2009, 54, 85–94. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhou, X.; Escala, W.; Papapetropoulos, S.; Zhai, R.G. β-N-Methylamino-L-Alanine Induces Neurological Deficits and Shortened Life Span in Drosophila. Toxins 2010, 2, 2663-2679. https://doi.org/10.3390/toxins2112663
Zhou X, Escala W, Papapetropoulos S, Zhai RG. β-N-Methylamino-L-Alanine Induces Neurological Deficits and Shortened Life Span in Drosophila. Toxins. 2010; 2(11):2663-2679. https://doi.org/10.3390/toxins2112663
Chicago/Turabian StyleZhou, Xianchong, Wilfredo Escala, Spyridon Papapetropoulos, and R. Grace Zhai. 2010. "β-N-Methylamino-L-Alanine Induces Neurological Deficits and Shortened Life Span in Drosophila" Toxins 2, no. 11: 2663-2679. https://doi.org/10.3390/toxins2112663
APA StyleZhou, X., Escala, W., Papapetropoulos, S., & Zhai, R. G. (2010). β-N-Methylamino-L-Alanine Induces Neurological Deficits and Shortened Life Span in Drosophila. Toxins, 2(11), 2663-2679. https://doi.org/10.3390/toxins2112663