Pharmacophore Selection and Redesign of Non-nucleotide Inhibitors of Anthrax Edema Factor

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pathway to Discovering a Family of Inhibitors of EF
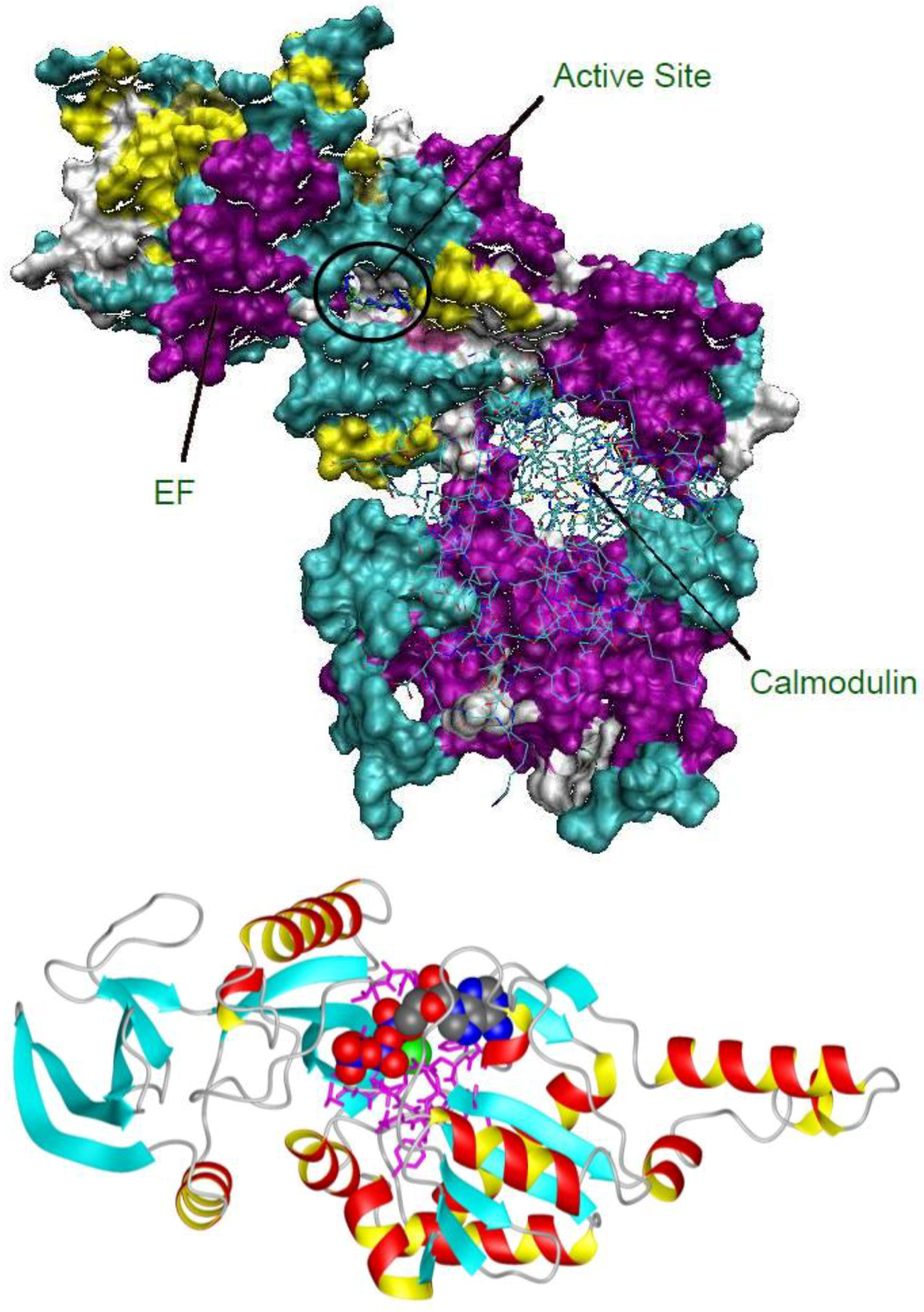
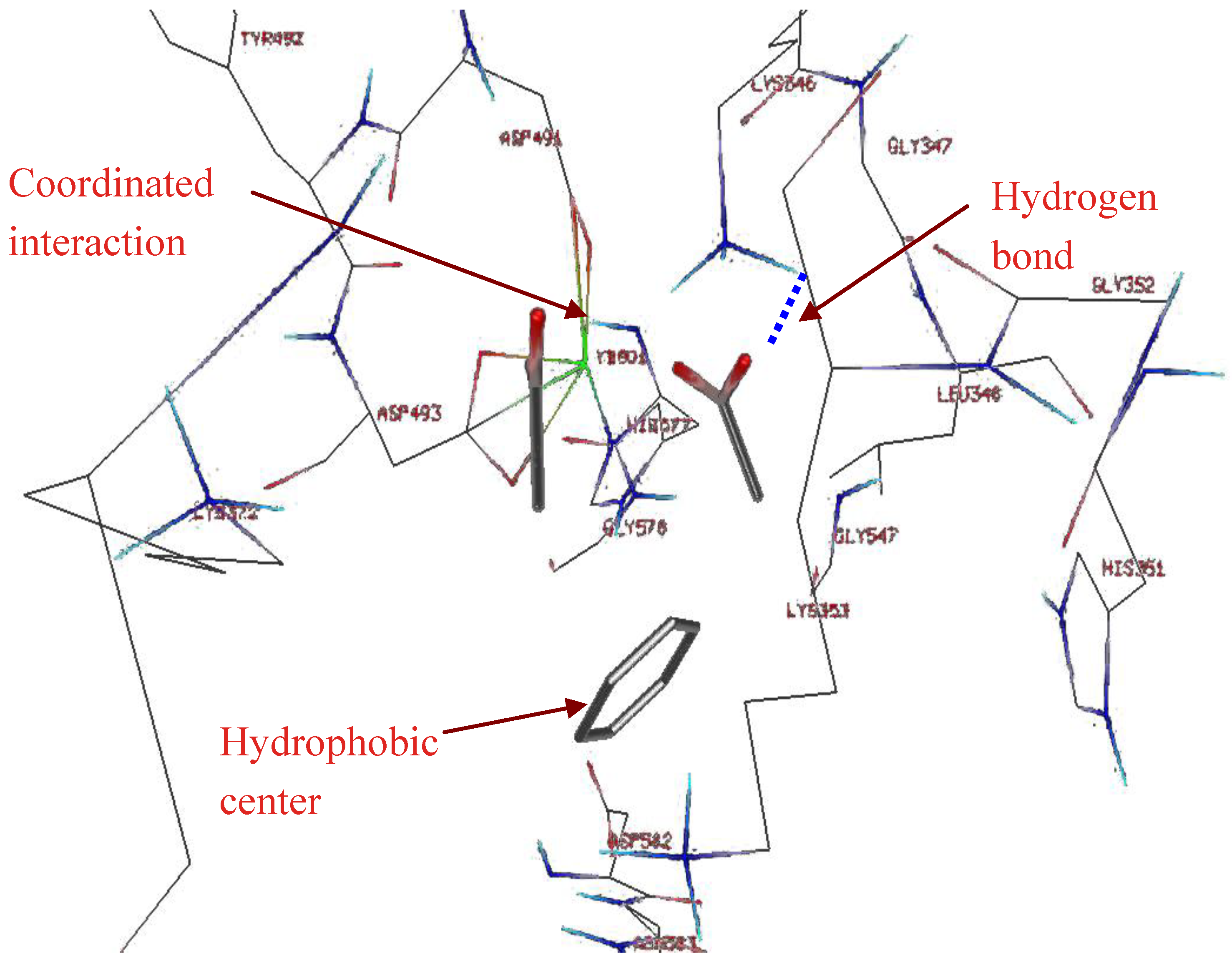
2.1. Studying the Active Site of EF


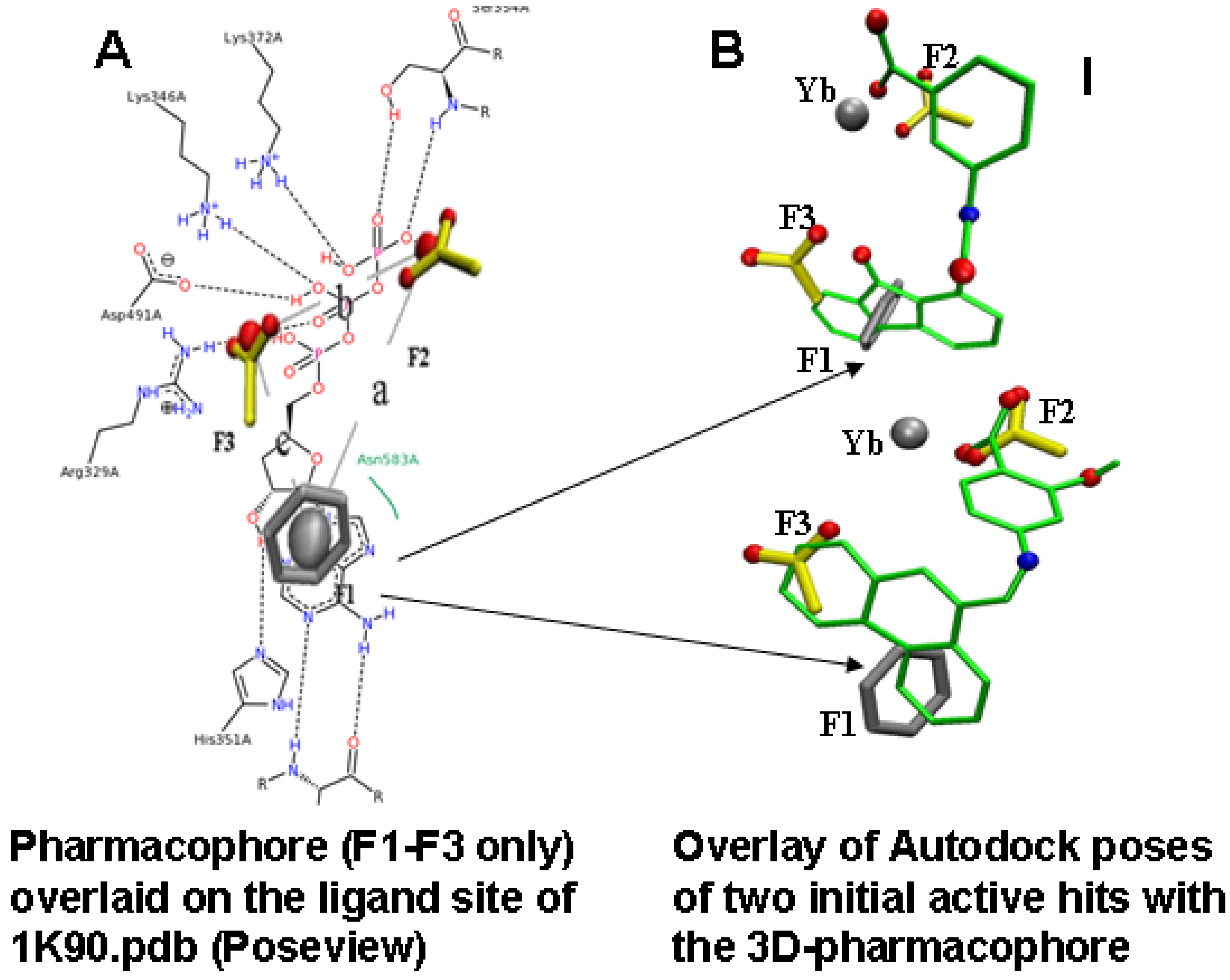
2.2. Compound Library Screening with a Fragment Based, 3D-Pharmacophore


2.3. Compound Selection Using Molecular Docking
3. Bioassay for Activities, Toxicities
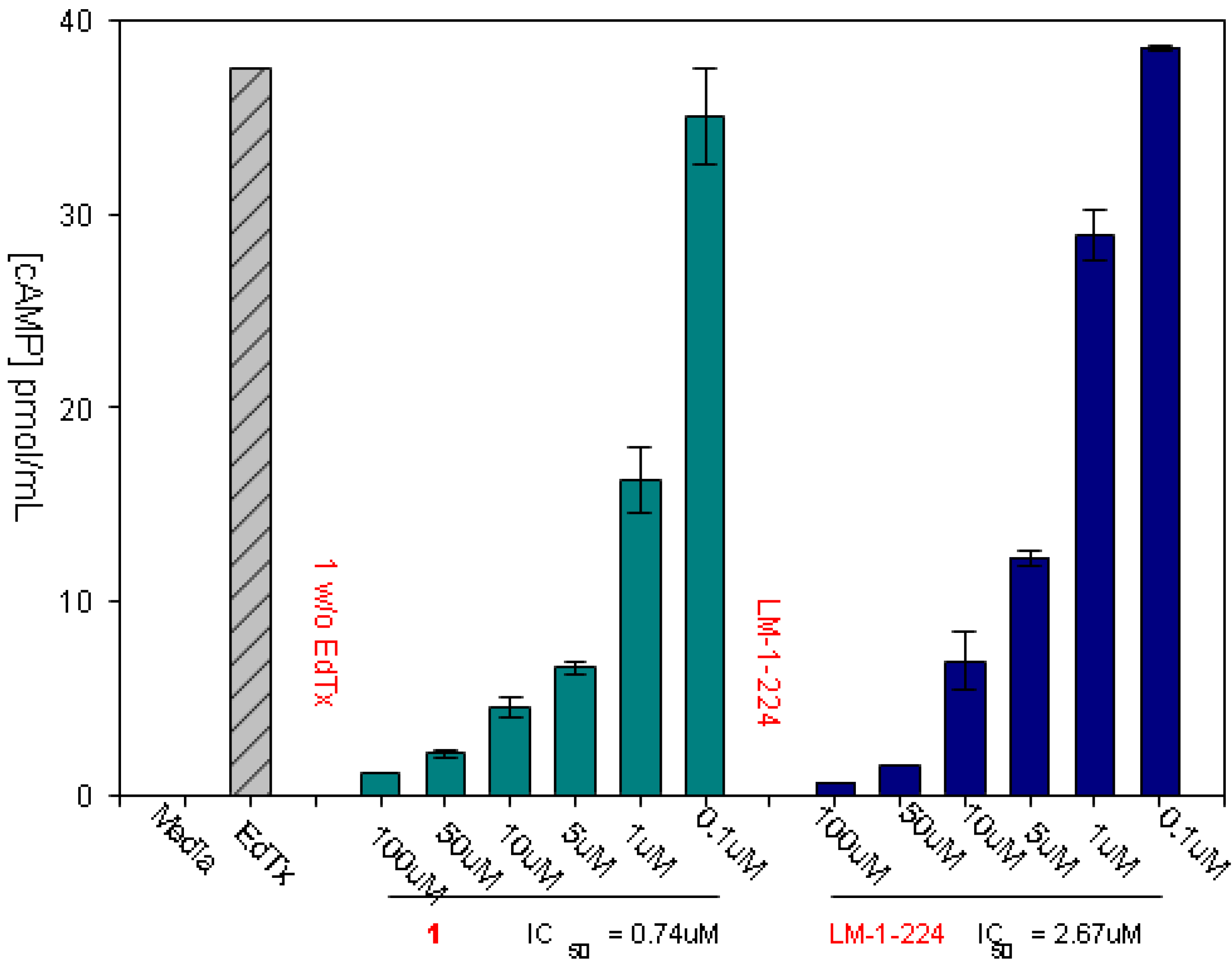
3.1. Experimental Assay for Inhibition of Toxin Induced Release of cAMP

3.2. Treating Enterotoxigenic E. coli (ETEC) Infections in a Murine Model
3.3. Additional Assays for Toxicity, and Estimation of Solubility
4. Redesign of the Inhibitors for Enhanced Solubility and Reduced Potential Toxicity
5. Conclusions
Acknowledgements
References and Notes
- Migone, T.S.; Subramanian, G.M.; Zhong, J.; Healey, L.M.; Corey, A.; Devalaraja, M.; Lo, L.; Ullrich, S.; Zimmerman, J.; Chen, A.; et al. Raxibacumab for the treatment of inhalational anthrax. N. Engl. J. Med. 2009, 361, 135–144. [Google Scholar]
- Sweeney, D.A.; Cui, X.; Solomon, S.B.; Vitberg, D.A.; Migone, T.S.; Scher, D.; Danner, R.L.; Natanson, C.; Subramanian, G.M.; Eichacker, P.Q. Anthrax lethal and edema toxins produce different patterns of cardiovascular and renal dysfunction and synergistically decrease survival in canines. J. Infect. Dis. 2010, 202, 1885–1896. [Google Scholar] [CrossRef]
- Lacy, D.B.; Collier, R.J. Structure and function of anthrax toxin. Curr. Top. Microbiol. Immunol. 2002, 271, 61–85. [Google Scholar]
- Guidi-Rontani, C.; Weber-Levy, M.; Mock, M.; Cabiaux, V. Translocation of Bacillus anthracis lethal and oedema factors across endosome membranes. Cell Microbiol. 2000, 2, 259–264. [Google Scholar] [CrossRef]
- Lacy, D.B.; Mourez, M.; Fouassier, A.; Collier, R.J. Mapping the anthrax protective antigen binding site on the lethal and edema factors. J. Biol. Chem. 2002, 277, 3006–3010. [Google Scholar]
- Peterson, J.W.; Comer, J.E.; Noffsinger, D.M.; Wenglikowski, A.; Walberg, K.G.; Chatuev, B.M.; Chopra, A.K.; Stanberry, L.R.; Kang, A.S.; Scholz, W.W.; Sircar, J. Human monoclonal anti-protective antigen antibody completely protects rabbits and is synergistic with ciprofloxacin in protecting mice and guinea pigs against inhalation anthrax. Infect. Immun. 2006, 74, 1016–1024. [Google Scholar]
- Abboud, N.; de Jesus, M.; Nakouzi, A.; Cordero, R.J.B.; Pujato, M.; Fiser, A.; Rivera, J.; Casadevall, A. Identification of linear epitopes in Bacillus anthracis protective antigen bound by neutralizing antibodies. J. Biol. Chem. 2009, 284, 25077–25086. [Google Scholar]
- Firoved, A.M.; Miller, G.F.; Moayeri, M.; Kakkar, R.; Shen, Y.; Wiggins, J.F.; McNally, E.M.; Tang, W.J.; Leppla, S.H. Bacillus anthracis edema toxin causes extensive tissue lesions and rapid lethality in mice. Am. J. Pathol. 2005, 167, 1309–1320. [Google Scholar] [CrossRef]
- Li, Q.; Peachman, K.K.; Sower, L.; Leppla, S.H.; Shivachandra, S.B.; Matyas, G.R.; Peterson, J.W.; Alving, C.R.; Rao, M.; Rao, V.B. Anthrax LFn-PA hybrid antigens: Biochemistry, immunogenicity, and protection against lethal ames spore challenge in rabbits. Open Vaccine J. 2009, 2, 92–99. [Google Scholar] [CrossRef]
- Lawrence, W.S.; Marshall, J.R.; Zavala, D.L.; Weaver, L.E.; Baze, W.B.; Moen, S.T.; Whorton, E.B.; Gourley, R.L.; Peterson, J.W. Hemodynamic effects of anthrax toxins in the rabbit model and the cardiac pathology induced by lethal toxin. Toxins 2011, 3, 721–736. [Google Scholar] [CrossRef]
- Park, H.C.; Sung, S.R.; Lim, S.M.; Lee, J.S.; Kim, S.K.; Yoon, M.Y. Proteolytic assay-based screening identifies a potent inhibitor of anthrax lethal factor. Microb. Pathog. 2012, 53, 109–112. [Google Scholar] [CrossRef]
- Li, F.; Terzyan, S.; Tang, J. Subsite specificity of anthrax lethal factor and its implications for inhibitor development. Biochem. Biophys. Res. Commun. 2011, 407, 400–405. [Google Scholar] [CrossRef]
- Bromberg-White, J.L.; Duesbery, N.S. Biological and biochemical characterization of anthrax lethal factor, a proteolytic inhibitor of MEK signaling pathways. Methods Enzymol. 2008, 438, 355–365. [Google Scholar]
- Taha, H.; Dove, S.; Geduhn, J.; König, B.; Shen, Y.; Tang, W.-J.; Seifert, R. Inhibition of the adenylyl cyclase toxin, edema factor, from Bacillus anthracis by a series of 18 mono- and bis-(M)ANT-substituted nucleoside 5'-triphosphates. Naunyn Schmiedebergs Arch. Pharmacol. 2012, 385, 57–68. [Google Scholar] [CrossRef]
- Laine, É.; Martínez, L.; Ladant, D.; Malliavin, T.; Blondel, A. Molecular motions as a drug target: Mechanistic simulations of anthrax toxin edema factor function led to the discovery of novel allosteric inhibitors. Toxins 2012, 4, 580–604. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Menche, G.; Power, T.D.; Sower, L.; Peterson, J.W.; Schein, C.H. Accounting for ligand-bound metal ions in docking small molecules on adenylyl cyclase toxins. Proteins 2007, 67, 593–605. [Google Scholar] [CrossRef]
- Drum, C.L.; Yan, S.Z.; Bard, J.; Shen, Y.Q.; Lu, D.; Soelaiman, S.; Grabarek, Z.; Bohm, A.; Tang, W.J. Structural basis for the activation of anthrax adenylyl cyclase exotoxin by calmodulin. Nature 2002, 415, 396–402. [Google Scholar]
- Fornabaio, M.; Cozzini, P.; Mozzarelli, A.; Abraham, D.J.; Kellogg, G.E. Simple, intuitive calculations of free energy of binding for protein-ligand complexes. 2. Computational titration and pH effects in molecular models of neuraminidase-inhibitor complexes. J. Med. Chem. 2003, 46, 4487–4500. [Google Scholar] [CrossRef]
- Fornabaio, M.; Spyrakis, F.; Mozzarelli, A.; Cozzini, P.; Abraham, D.J.; Kellogg, G.E. Simple, intuitive calculations of free energy of binding for protein-ligand complexes. 3. The free energy contribution of structural water molecules in HIV-1 protease complexes. J. Med. Chem. 2004, 47, 4507–4516. [Google Scholar] [CrossRef]
- Cozzini, P.; Fornabaio, M.; Marabotti, A.; Abraham, D.J.; Kellogg, G.E.; Mozzarelli, A. Simple, intuitive calculations of free energy of binding for protein-ligand complexes. 1. Models without explicit constrained water. J. Med. Chem. 2002, 45, 2469–2483. [Google Scholar] [CrossRef]
- Stierand, K.; Rarey, M. Drawing the PDB: Protein-ligand complexes in two dimensions. ACS Med. Chem. Lett. 2010, 1, 540–545. [Google Scholar] [CrossRef]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef]
- Compound 1 is referred to in our patent (reference 35) as FIV-50; it was also called DC-5 in our earlier publications. LM-1-224 was called FIV-35 in our patent (reference 36), and is compound 9 in reference 37. The reader is referred to the patent and paper for more details about active derivatives.
- Chen, D.; Misra, M.; Sower, L.; Peterson, J.W.; Kellogg, G.E.; Schein, C.H. Novel inhibitors of anthrax edema factor. Bioorg. Med. Chem. 2008, 16, 7225–7233. [Google Scholar]
- Natesan, S.; Subramaniam, R.; Bergeron, C.; Balaz, S. Binding affinity prediction for ligands and receptors forming tautomers and ionization species: Inhibition of mitogen-activated protein kinase-activated protein kinase 2 (MK2). J. Med. Chem. 2012, 55, 2035–2047. [Google Scholar] [CrossRef]
- Moen, S.T.; Blumentritt, C.A.; Slater, T.M.; Patel, S.D.; Tutt, C.B.; Estrella-Jimenez, M.E.; Pawlik, J.; Sower, L.; Popov, V.L.; Schein, C.H.; et al. Testing the efficacy and toxicity of adenylyl cyclase inhibitors against enteric pathogens using in vitro and in vivo models of infection. Infect. Immun. 2010, 78, 1740–1749. [Google Scholar] [CrossRef]
- Wang, X.; Gao, X.; Hardwidge, P.R. Heat-labile enterotoxin-induced activation of NF-kappaB and MAPK pathways in intestinal epithelial cells impacts enterotoxigenic Escherichia coli (ETEC) adherence. Cell. Microbiol. 2012, 14, 1231–1241. [Google Scholar] [CrossRef]
- Flores, J.; DuPont, H.L.; Lee, S.A.; Belkind-Gerson, J.; Paredes, M.; Mohamed, J.A.; Armitige, L.Y.; Guo, D.C.; Okhuysen, P.C. Influence of host interleukin-10 polymorphisms on development of traveler’s diarrhea due to heat-labile enterotoxin-producing Escherichia coli in travelers from the United States who are visiting Mexico. Clin. Vaccine Immunol. 2008, 15, 1194–1198. [Google Scholar] [CrossRef]
- Daniels, N.A. Enterotoxigenic Escherichia coli: Traveler’s diarrhea comes home. Clin. Infect. Dis. 2006, 42, 335–336. [Google Scholar] [CrossRef]
- Tobias, J.; Svennerholm, A.M. Strategies to overexpress enterotoxigenic Escherichia coli (ETEC) colonization factors for the construction of oral whole-cell inactivated ETEC vaccine candidates. Appl. Microbiol. Biotechnol. 2012, 93, 2291–2300. [Google Scholar] [CrossRef]
- Beatty, M.E.; Adcock, P.M.; Smith, S.W.; Quinlan, K.; Kamimoto, L.A.; Rowe, S.Y.; Scott, K.; Conover, C.; Varchmin, T.; Bopp, C.A.; et al. Epidemic diarrhea due to enterotoxigenic Escherichia coli. Clin. Infect. Dis. 2006, 42, 329–334. [Google Scholar] [CrossRef]
- Sahl, J.W.; Steinsland, H.; Redman, J.C.; Angiuoli, S.V.; Nataro, J.P.; Sommerfelt, H.; Rasko, D.A. A comparative genomic analysis of diverse clonal types of enterotoxigenic Escherichia coli reveals pathovar-specific conservation. Infect. Immun. 2011, 79, 950–960. [Google Scholar] [CrossRef]
- Liang, W.; Pascual-Montano, A.; Silva, A.J.; Benitez, J.A. The cyclic AMP receptor protein modulates quorum sensing, motility and multiple genes that affect intestinal colonization in Vibrio cholerae. Microbiology 2007, 153, 2964–2975. [Google Scholar] [CrossRef]
- Surette, M.G.; Miller, M.B.; Bassler, B.L. Quorum sensing in Escherichia coli, Salmonella typhimurium, and Vibrio harveyi: A new family of genes responsible for autoinducer production. Proc. Natl. Acad. Sci. 1999, 96, 1639–1644. [Google Scholar] [CrossRef]
- Chen, D.; Martin, Z.S.; Soto, C.; Schein, C.H. Computational selection of inhibitors of Abeta aggregation and neuronal toxicity. Bioorg. Med. Chem. 2009, 17, 5189–5197. [Google Scholar] [CrossRef]
- Schein, C.H.; Chen, D.; Gilbertson, S.R.; Estrella-Jimenes, M.; Gao, J.; Walter, M.A.; Peterson, J. W. Methods and Compositions to Inhibit Edema Factor and Adenylyl Cyclase. U.S. Patent 8003692, 23 August 2011. [Google Scholar]
- Chen, D.; Ma, L.; Kanalas, J.J.; Gao, J.; Pawlik, J.; Jimenez, M.E.; Walter, M.A.; Peterson, J.W.; Gilbertson, S.R.; Schein, C.H. Structure-based redesign of an edema toxin inhibitor. Bioorg. Med. Chem. 2012, 20, 368–376. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Huey, R.; Olson, A.J. Distributed automated docking of flexible ligands to proteins: Parallel applications of AutoDock 2.4. J. Comput. Aided Mol. Des. 1996, 10, 293–304. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Schein, C.H.; Chen, D.; Ma, L.; Kanalas, J.J.; Gao, J.; Jimenez, M.E.; Sower, L.E.; Walter, M.A.; Gilbertson, S.R.; Peterson, J.W. Pharmacophore Selection and Redesign of Non-nucleotide Inhibitors of Anthrax Edema Factor. Toxins 2012, 4, 1288-1300. https://doi.org/10.3390/toxins4111288
Schein CH, Chen D, Ma L, Kanalas JJ, Gao J, Jimenez ME, Sower LE, Walter MA, Gilbertson SR, Peterson JW. Pharmacophore Selection and Redesign of Non-nucleotide Inhibitors of Anthrax Edema Factor. Toxins. 2012; 4(11):1288-1300. https://doi.org/10.3390/toxins4111288
Chicago/Turabian StyleSchein, Catherine H., Deliang Chen, Lili Ma, John J. Kanalas, Jian Gao, Maria Estrella Jimenez, Laurie E. Sower, Mary A. Walter, Scott R. Gilbertson, and Johnny W. Peterson. 2012. "Pharmacophore Selection and Redesign of Non-nucleotide Inhibitors of Anthrax Edema Factor" Toxins 4, no. 11: 1288-1300. https://doi.org/10.3390/toxins4111288
APA StyleSchein, C. H., Chen, D., Ma, L., Kanalas, J. J., Gao, J., Jimenez, M. E., Sower, L. E., Walter, M. A., Gilbertson, S. R., & Peterson, J. W. (2012). Pharmacophore Selection and Redesign of Non-nucleotide Inhibitors of Anthrax Edema Factor. Toxins, 4(11), 1288-1300. https://doi.org/10.3390/toxins4111288