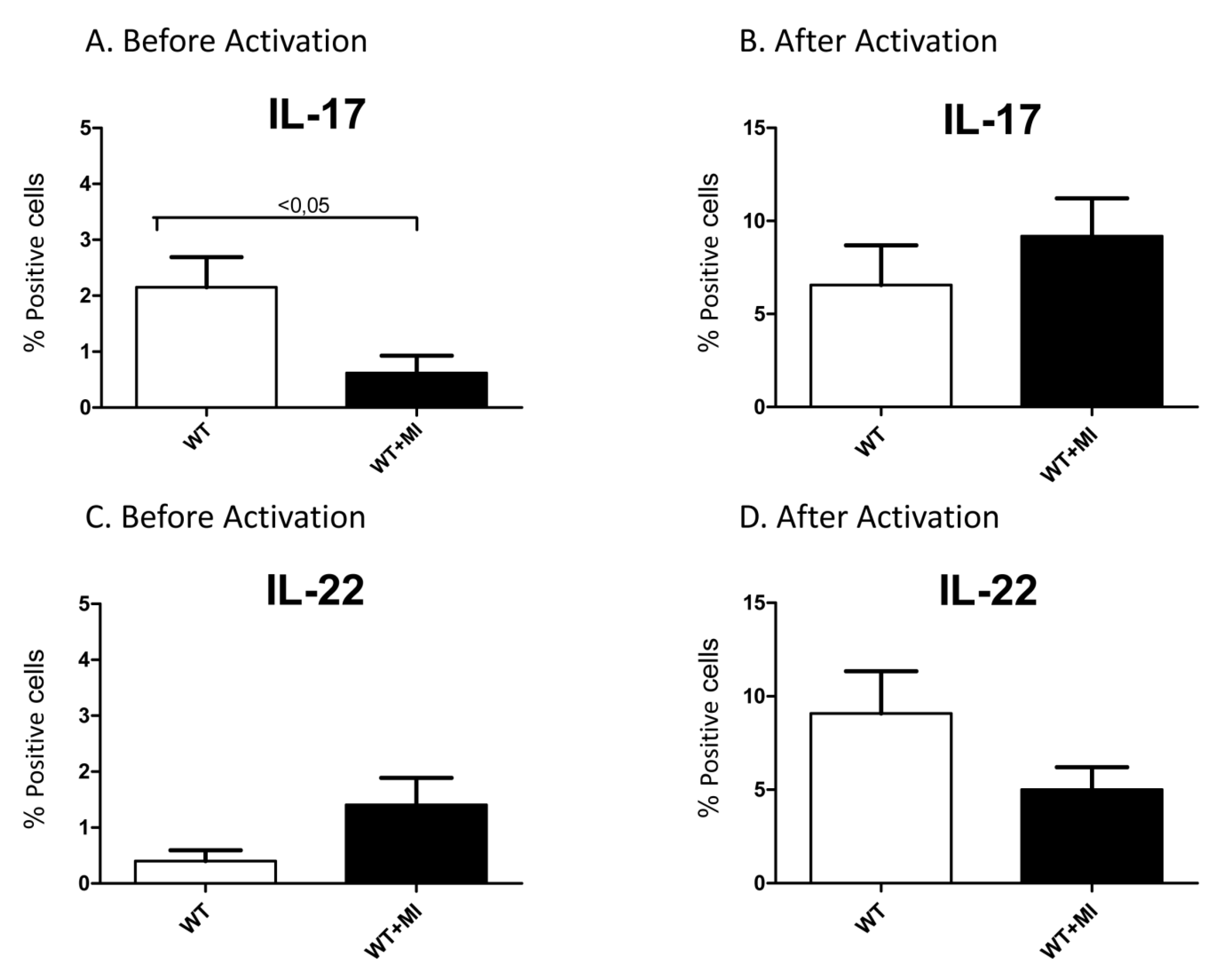
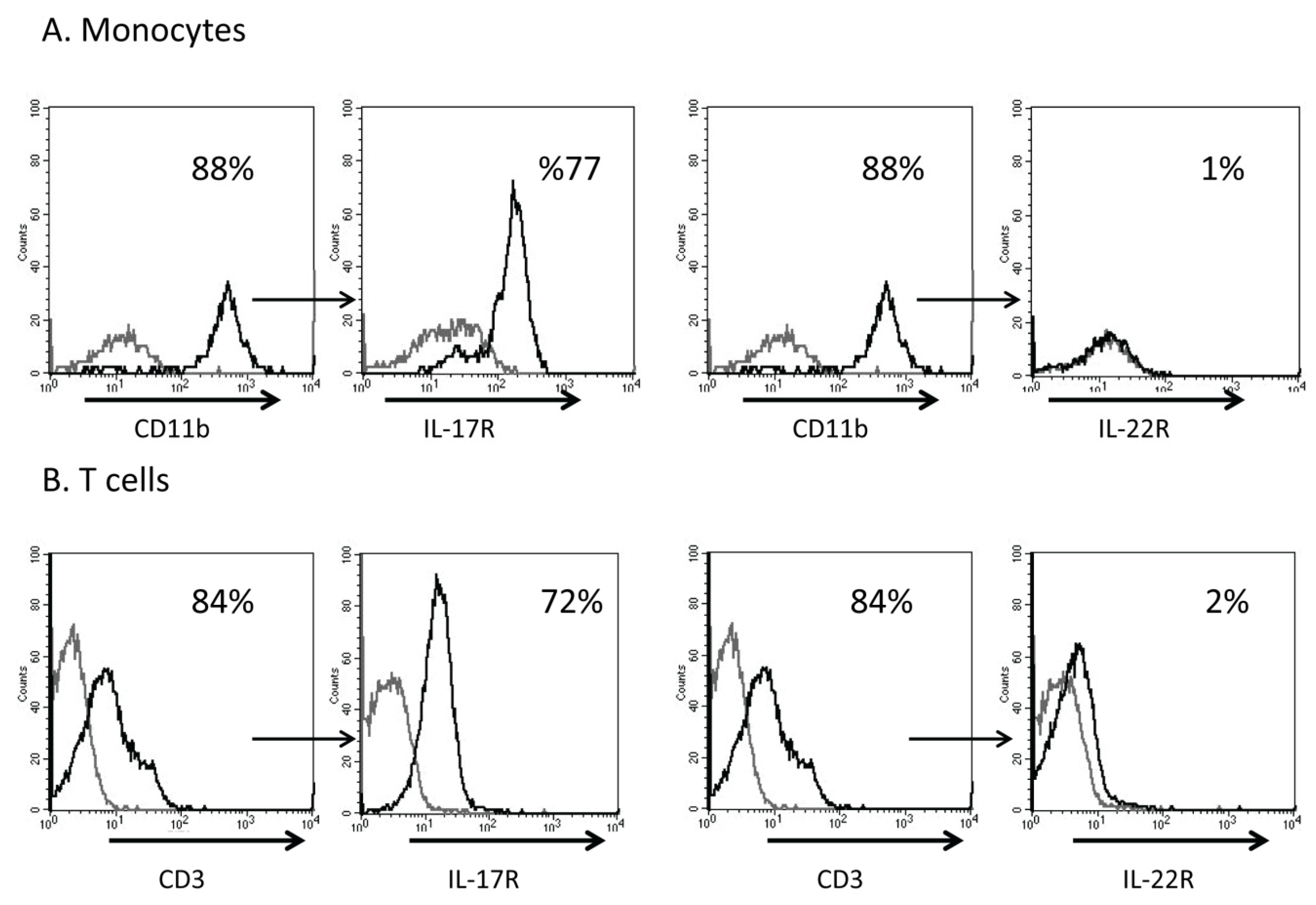
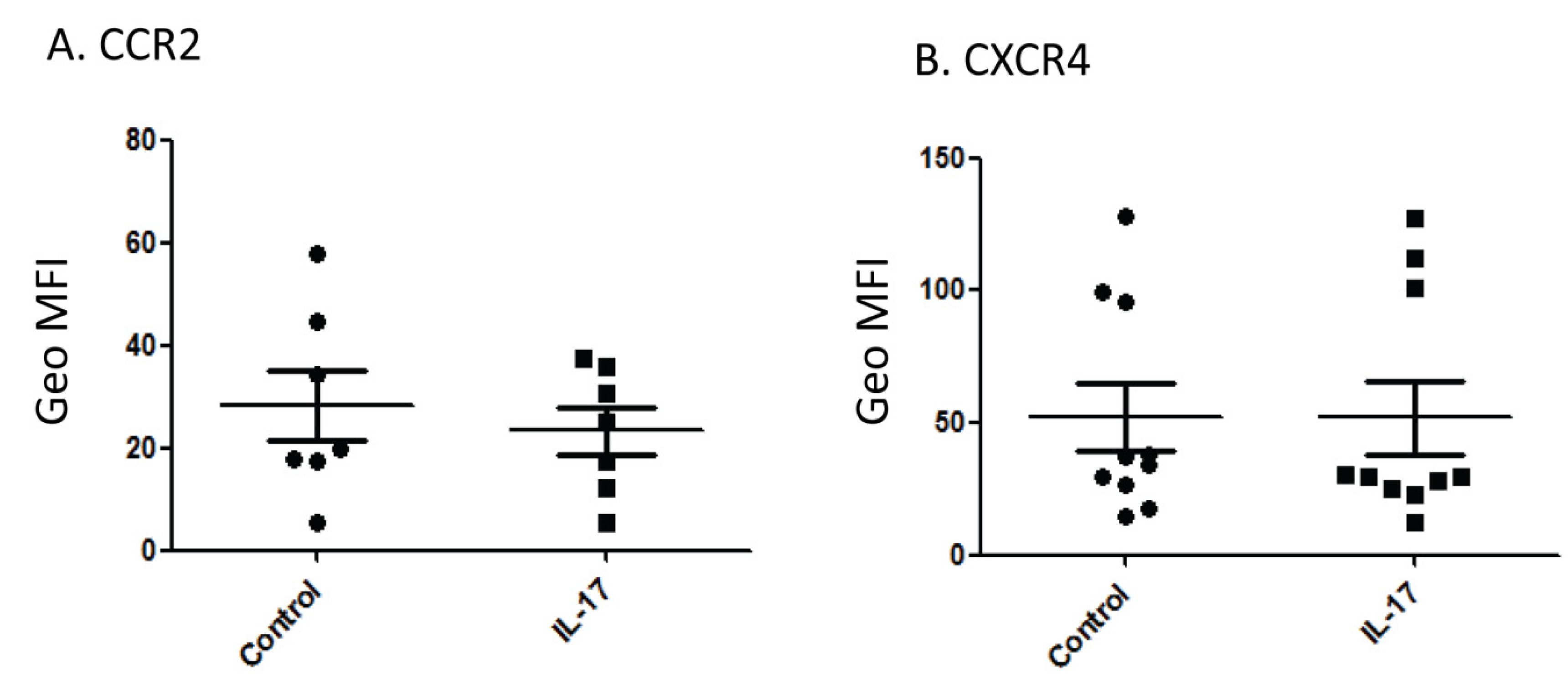
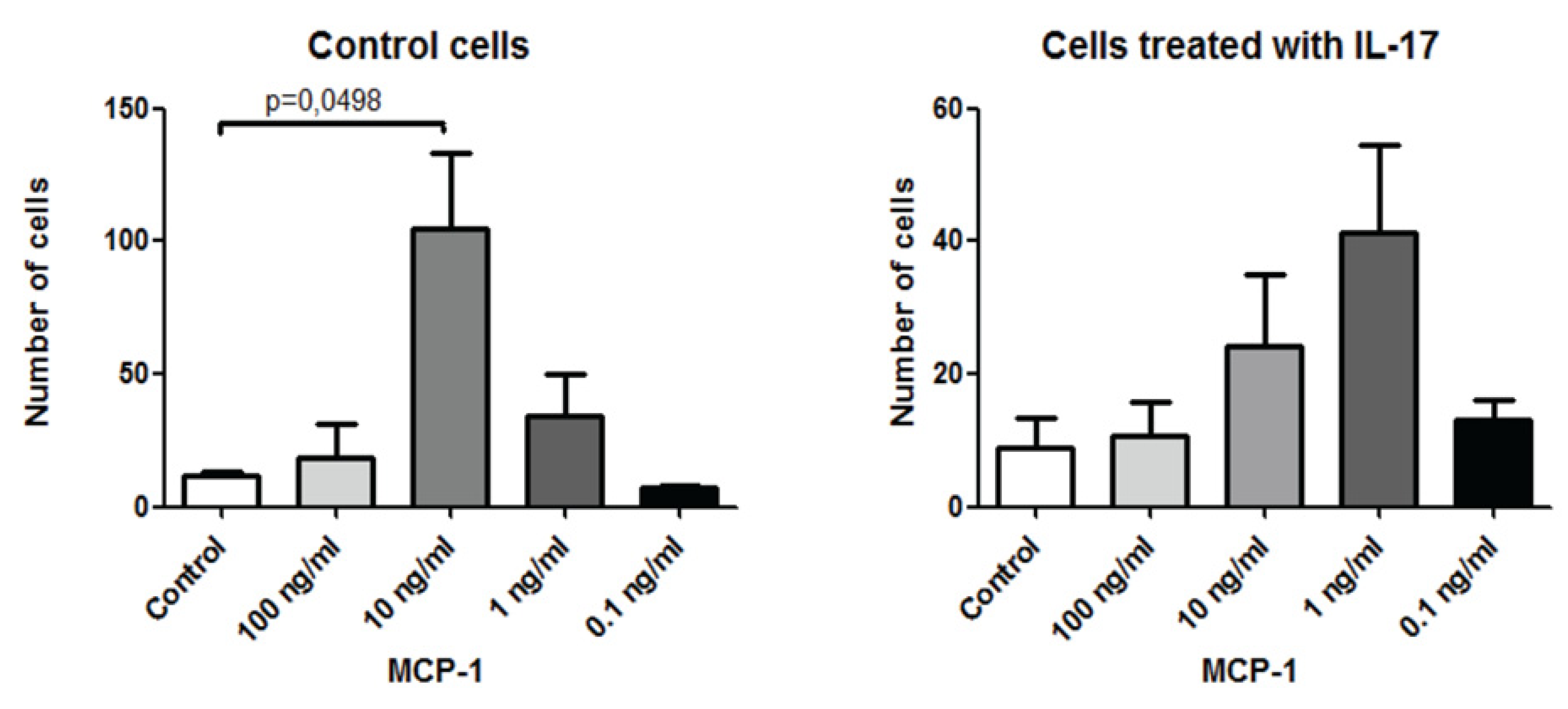
Interleukin-17 (IL-17) Expression Is Reduced during Acute Myocardial Infarction: Role on Chemokine Receptor Expression in Monocytes and Their in Vitro Chemotaxis towards Chemokines


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
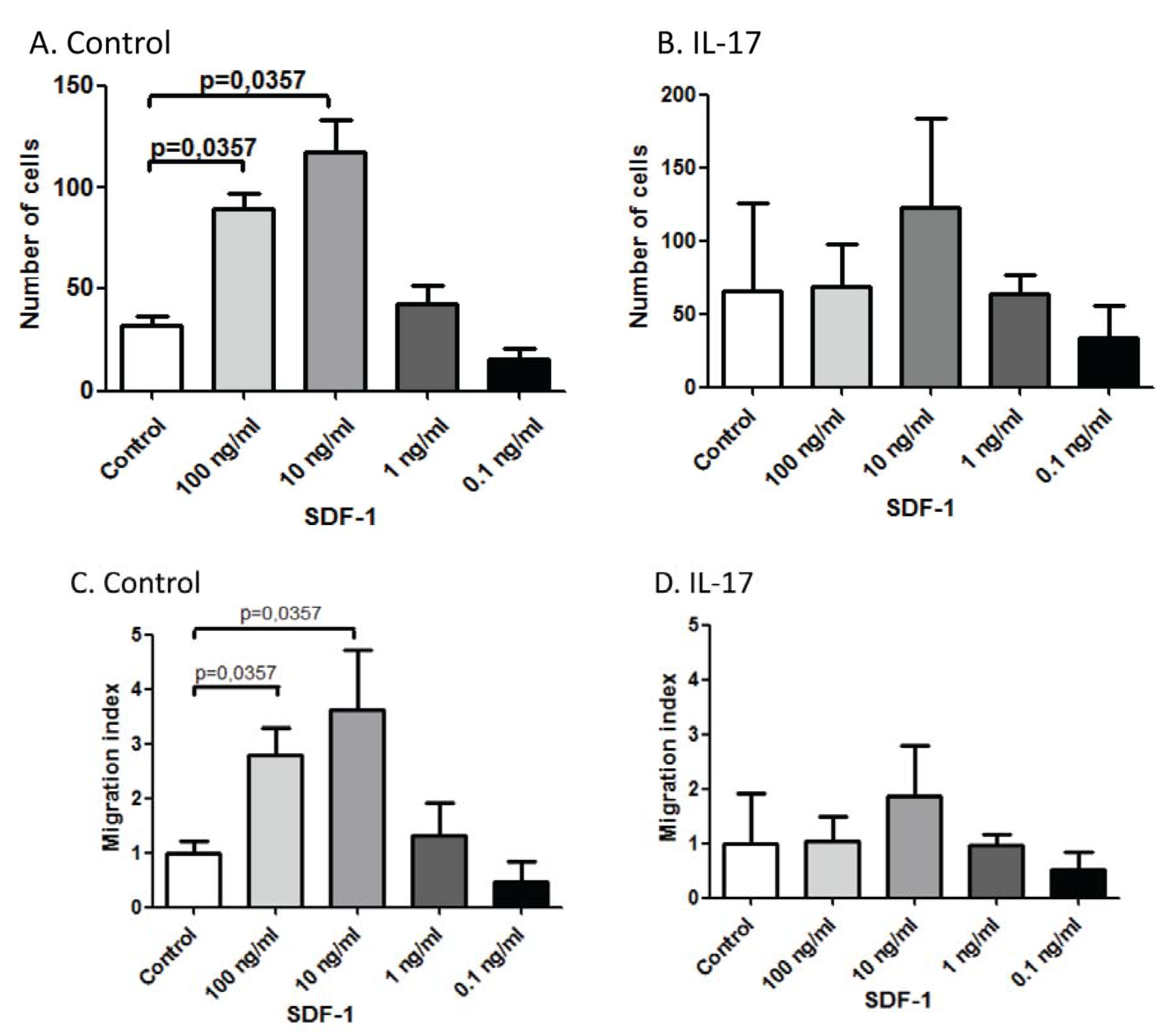
Abstract
Share and Cite
Troitskaya, M.; Baysa, A.; Vaage, J.; Sand, K.L.; Maghazachi, A.A.; Valen, G. Interleukin-17 (IL-17) Expression Is Reduced during Acute Myocardial Infarction: Role on Chemokine Receptor Expression in Monocytes and Their in Vitro Chemotaxis towards Chemokines. Toxins 2012, 4, 1427-1439. https://doi.org/10.3390/toxins4121427
Troitskaya M, Baysa A, Vaage J, Sand KL, Maghazachi AA, Valen G. Interleukin-17 (IL-17) Expression Is Reduced during Acute Myocardial Infarction: Role on Chemokine Receptor Expression in Monocytes and Their in Vitro Chemotaxis towards Chemokines. Toxins. 2012; 4(12):1427-1439. https://doi.org/10.3390/toxins4121427
Chicago/Turabian StyleTroitskaya, Maria, Anton Baysa, Jarle Vaage, Kristin L. Sand, Azzam A. Maghazachi, and Guro Valen. 2012. "Interleukin-17 (IL-17) Expression Is Reduced during Acute Myocardial Infarction: Role on Chemokine Receptor Expression in Monocytes and Their in Vitro Chemotaxis towards Chemokines" Toxins 4, no. 12: 1427-1439. https://doi.org/10.3390/toxins4121427
APA StyleTroitskaya, M., Baysa, A., Vaage, J., Sand, K. L., Maghazachi, A. A., & Valen, G. (2012). Interleukin-17 (IL-17) Expression Is Reduced during Acute Myocardial Infarction: Role on Chemokine Receptor Expression in Monocytes and Their in Vitro Chemotaxis towards Chemokines. Toxins, 4(12), 1427-1439. https://doi.org/10.3390/toxins4121427