Bacillus anthracis Edema Factor Substrate Specificity: Evidence for New Modes of Action

Abstract
:Abbreviations:
| AC | adenylyl cyclase |
| ACD | adenylyl cyclase domain |
| ANT | anthraniloyl |
| EF | edema factor AC toxin from Bacillus anthracis |
| CaM | calmodulin |
| cAMP | cyclic adenosine 3′:5′-monophosphate |
| ANTXR | anthrax toxin receptor |
| CC | cytidylyl cyclase |
| cCMP | cyclic cytidine 3′:5′-monophosphate |
| cIMP | cyclic inosine 3′:5′-monophosphate |
| CMG2 | capillary morphogenesis gene 2 |
| cNMP | cyclic nucleoside 3′:5′-monophosphate |
| CREB | cAMP response element-binding |
| cUMP | cyclic uridine 3′:5′-monophosphate |
| cXMP | cyclic xanthosine 3′:5′-monophosphate |
| IBMX | 3-isobutyl-1-methylxanthine |
| IC | inosylyl cyclase |
| LF | lethal factor |
| mAC | membranous mammalian AC |
| MAPK | mitogen-activated protein kinase |
| MAPKK | mitogen-activated protein kinase kinase |
| MANT | methylanthraniloyl |
| MRP | multidrug resistance protein |
| MS | mass spectrometry |
| NTP | nucleoside 5′-triphosphate |
| PA | protective antigen |
| PDE | phosphodiesterase |
| PKA | cAMP-dependent protein kinase |
| PKG | cGMP-dependent protein kinase |
| PMEApp | 9-[2-(phosphonomethoxy)ethyl]adenine diphosphate |
| PMN | polymorphonuclear leukocytes |
| sGC | soluble mammalian guanylyl cyclase |
| TEM | transendothelial migration |
| TEM8 | tumor endothelial marker 8 |
| UC | uridylyl cyclase |
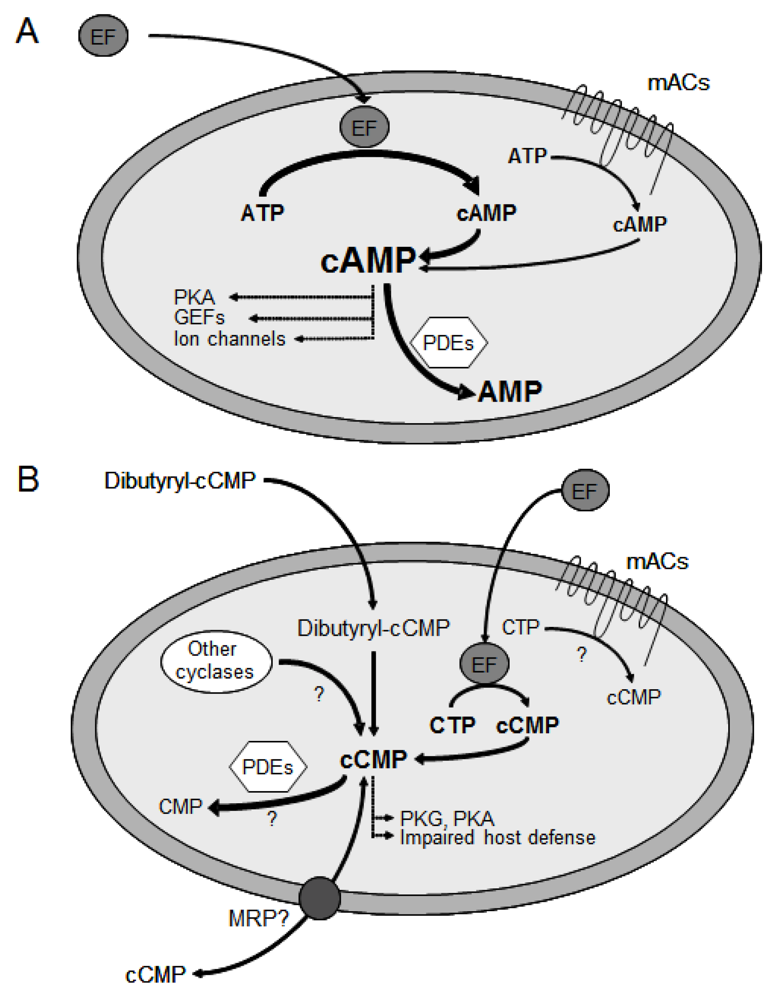
1. The cAMP Signaling Pathway

2. The Fatal Synergism of Bacillus anthracis Exotoxin Components


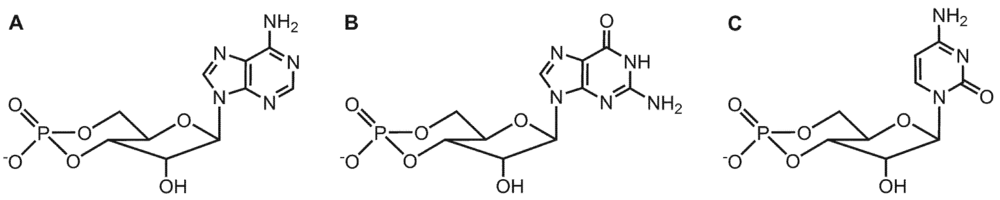
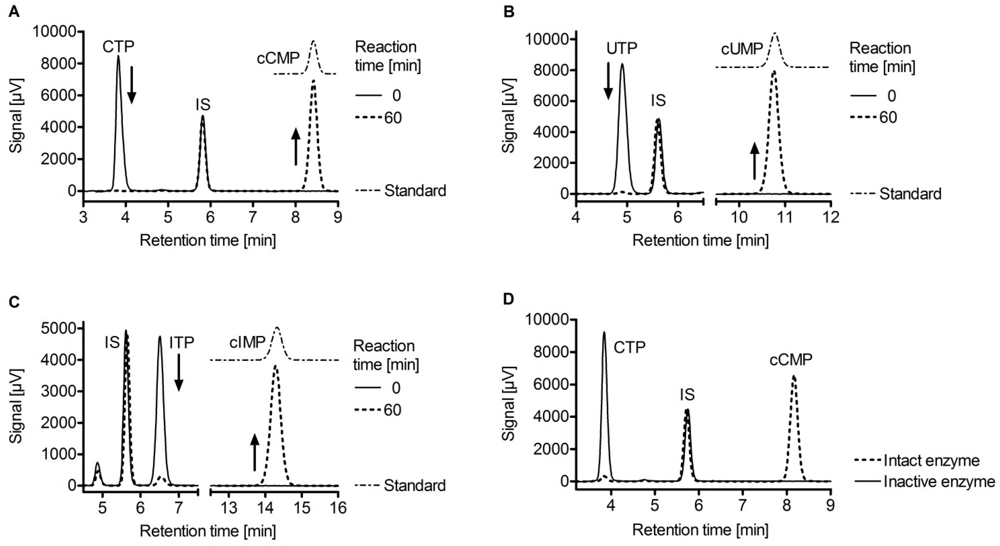
3. Substrate-Specificity of EF: cCMP, cUMP and cIMP as Potential New Second Messengers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | AC 1 (nM) | AC 2 (nM) | AC 5 (nM) | EF (nM) |
|---|---|---|---|---|
| MANT-ATP | 150 | 330 | 100 | 580 |
| MANT-ITP | 2.8 | 14 | 1.2 | 4,100 |
| MANT-GTP | 90 | 620 | 55 | 2,500 |
| MANT-CTP | 150 | 690 | 150 | 100 |
| MANT-UTP | 46 | 460 | 32 | 3,700 |

| Enzyme | NC activity | Me2+ | Km [µM] | kcat [s-1] |
|---|---|---|---|---|
| EF | AC | Mn2+ | 35.3 ± 3.7 | 501.5 ± 55.9 |
| Mg2+ | 175.8 ± 29.9 | 684.2 ± 272.5 | ||
| CC | Mn2+ | 12.5 ± 3.4 | 8.8 ± 1.4 | |
| Mg2+ | 419.7 ± 115.1 | 7.2 ± 3.1 | ||
| UC | Mn2+ | 134.5 ± 23.5 | 2.3 ± 0.2 |
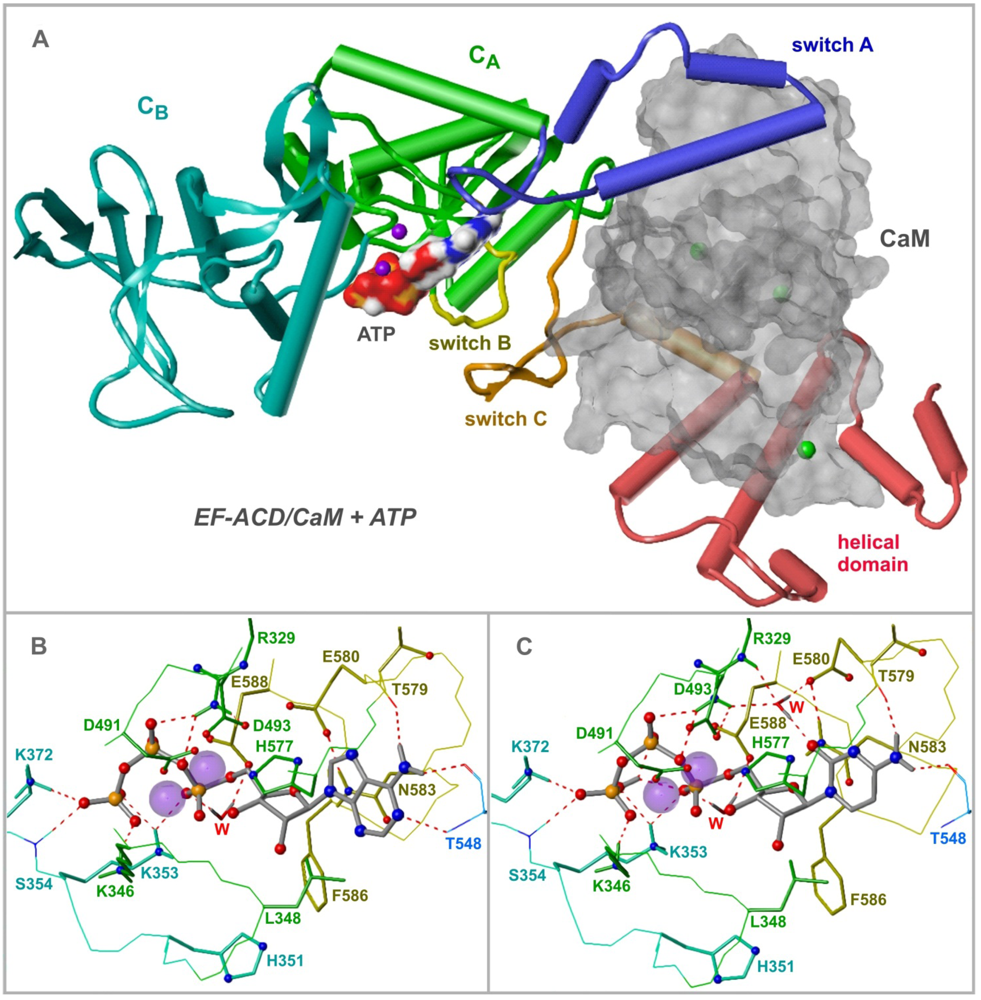
4. EF Structure and Nucleotide Binding Modes

5. Potential Cellular Targets of Novel Cyclic Nucleotides: Protein Kinases, Phosphodiesterases and Cyclic Nucleotide-Gated Ion Channels
5.1. Protein Kinases (PKs)
5.2. Cyclic Nucleotide Phosphodiesterases (PDEs)
5.3. Cyclic Nucleotide-Gated Ion Channels (CNGs)
6. Unresolved Questions and Future Studies
7. Conclusions
Acknowledgements
Conflict of Interest
References
- Mosenden, R.; Tasken, K. Cyclic AMP-mediated immune regulation-overview of mechanisms of action in T cells. Cell. Signal. 2011, 23, 1009–1016. [Google Scholar] [CrossRef]
- Defer, N.; Best-Belpomme, M.; Hanoune, J. Tissue specificity and physiological relevance of various isoforms of adenylyl cyclase. Am. J. Physiol. Renal Physiol. 2000, 279, F400–F416. [Google Scholar]
- Sadana, R.; Dessauer, C.W. Physiological roles for G protein-regulated adenylyl cyclase isoforms: Insights from knockout and overexpression studies. Neurosignals 2009, 17, 5–22. [Google Scholar] [CrossRef]
- Chen, J.; Levin, L.R.; Buck, J. Role of soluble adenylyl cyclase in the heart. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H538–543. [Google Scholar] [CrossRef]
- Hanoune, J.; Defer, N. Regulation and role of adenylyl cyclase isoforms. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 145–174. [Google Scholar] [CrossRef]
- Hanoune, J.; Pouille, Y.; Tzavara, E.; Shen, T.; Lipskaya, L.; Miyamoto, N.; Suzuki, Y.; Defer, N. Adenylyl cyclases: Structure, regulation and function in an enzyme superfamily. Mol. Cell. Endocrinol. 1997, 128, 179–194. [Google Scholar] [CrossRef]
- Patel, T.B.; Du, Z.; Pierre, S.; Cartin, L.; Scholich, K. Molecular biological approaches to unravel adenylyl cyclase signaling and function. Gene 2001, 269, 13–25. [Google Scholar] [CrossRef]
- Shirshev, S.V. Role of Epac proteins in mechanisms of cAMP-dependent immunoregulation. Biochemistry (Moscow) 2011, 76, 981–998. [Google Scholar]
- Breckler, M.; Berthouze, M.; Laurent, A.C.; Crozatier, B.; Morel, E.; Lezoualc'h, F. Rap-linked cAMP signaling Epac proteins: compartmentation, functioning and disease implications. Cell. Signal. 2011, 23, 1257–1266. [Google Scholar] [CrossRef]
- Laurent, A.C.; Breckler, M.; Berthouze, M.; Lezoualc'h, F. Role of Epac in brain and heart. Biochem. Soc. Trans. 2012, 40, 51–57. [Google Scholar] [CrossRef]
- Kleppe, R.; Krakstad, C.; Selheim, F.; Kopperud, R.; Doskeland, S.O. The cAMP-dependent protein kinase pathway as therapeutic target: Possibilities and pitfalls. Curr. Top. Med. Chem. 2011, 11, 1393–1405. [Google Scholar] [CrossRef]
- Göttle, M.; Geduhn, J.; König, B.; Gille, A.; Höcherl, K.; Seifert, R. Characterization of mouse heart adenylyl cyclase. J. Pharmacol. Exp. Ther. 2009, 329, 1156–1165. [Google Scholar] [CrossRef]
- Stangherlin, A.; Zaccolo, M. cGMP-cAMP interplay in cardiac myocytes: A local affair with far-reaching consequences for heart function. Biochem. Soc. Trans. 2012, 40, 11–14. [Google Scholar] [CrossRef]
- Morgado, M.; Cairrao, E.; Santos-Silva, A.J.; Verde, I. Cyclic nucleotide-dependent relaxation pathways in vascular smooth muscle. Cell. Mol. Life Sci. 2012, 69, 247–266. [Google Scholar] [CrossRef]
- Potter, L.R. Guanylyl cyclase structure, function and regulation. Cell. Signal. 2011, 23, 1921–1926. [Google Scholar] [CrossRef]
- Hammond, J.; Balligand, J.L. Nitric oxide synthase and cyclic GMP signaling in cardiac myocytes: From contractility to remodeling. J. Mol. Cell. Cardiol. 2012, 52, 330–340. [Google Scholar] [CrossRef]
- Francis, S.H.; Blount, M.A.; Corbin, J.D. Mammalian cyclic nucleotide phosphodiesterases: Molecular mechanisms and physiological functions. Physiol. Rev. 2011, 91, 651–690. [Google Scholar] [CrossRef]
- Russel, F.G.; Koenderink, J.B.; Masereeuw, R. Multidrug resistance protein 4 (MRP4/ABCC4): A versatile efflux transporter for drugs and signalling molecules. Trends Pharmacol. Sci. 2008, 29, 200–207. [Google Scholar] [CrossRef]
- Copsel, S.; Garcia, C.; Diez, F.; Vermeulem, M.; Baldi, A.; Bianciotti, L.G.; Russel, F.G.; Shayo, C.; Davio, C. Multidrug resistance protein 4 (MRP4/ABCC4) regulates cAMP cellular levels and controls human leukemia cell proliferation and differentiation. J. Biol. Chem. 2011, 286, 6979–6988. [Google Scholar]
- Ravna, A.W.; Sylte, I.; Sager, G. A molecular model of a putative substrate releasing conformation of multidrug resistance protein 5 (MRP5). Eur. J. Med. Chem. 2008, 43, 2557–2567. [Google Scholar] [CrossRef]
- McDonough, K.A.; Rodriguez, A. The myriad roles of cyclic AMP in microbial pathogens: From signal to sword. Nat. Rev. Microbiol. 2012, 10, 27–38. [Google Scholar]
- Vojtova, J.; Kamanova, J.; Sebo, P. Bordetella adenylate cyclase toxin: A swift saboteur of host defense. Curr. Opin. Microbiol. 2006, 9, 69–75. [Google Scholar]
- Ahuja, N.; Kumar, P.; Bhatnagar, R. The adenylate cyclase toxins. Crit. Rev. Microbiol. 2004, 30, 187–196. [Google Scholar] [CrossRef]
- Carbonetti, N.H.; Artamonova, G.V.; Andreasen, C.; Bushar, N. Pertussis toxin and adenylate cyclase toxin provide a one-two punch for establishment of Bordetella pertussis infection of the respiratory tract. Infect. Immun. 2005, 73, 2698–2703. [Google Scholar]
- Carbonetti, N.H.; Artamonova, G.V.; Mays, R.M.; Worthington, Z.E. Pertussis toxin plays an early role in respiratory tract colonization by Bordetella pertussis. Infect. Immun. 2003, 71, 6358–6366. [Google Scholar]
- Abramowitz, J.; Campbell, A.R. Cholera toxin action on rabbit corpus luteum membranes: Effects on adenylyl cyclase activity and adenosine diphospho-ribosylation of the stimulatory guanine nucleotide-binding regulatory component. Biol. Reprod. 1985, 32, 463–474. [Google Scholar] [CrossRef]
- Ivarsson, M.E.; Leroux, J.C.; Castagner, B. Targeting bacterial toxins. Angew. Chem. Int. Ed. Engl. 2012, 51, 4024–4045. [Google Scholar]
- Tang, W.J.; Guo, Q. The adenylyl cyclase activity of anthrax edema factor. Mol. Aspects Med. 2009, 30, 423–430. [Google Scholar] [CrossRef]
- Tournier, J.N.; Rossi Paccani, S.; Quesnel-Hellmann, A.; Baldari, C.T. Anthrax toxins: A weapon to systematically dismantle the host immune defenses. Mol. Aspects Med. 2009, 30, 456–466. [Google Scholar] [CrossRef]
- Rossi Paccani, S.; Benagiano, M.; Capitani, N.; Zornetta, I.; Ladant, D.; Montecucco, C.; D'Elios, M.M.; Baldari, C.T. The adenylate cyclase toxins of Bacillus anthracis and Bordetella pertussis promote Th2 cell development by shaping T cell antigen receptor signaling. PLoS Pathog. 2009, 5, e1000325. [Google Scholar] [CrossRef]
- Bauche, C.; Chenal, A.; Knapp, O.; Bodenreider, C.; Benz, R.; Chaffotte, A.; Ladant, D. Structural and functional characterization of an essential RTX subdomain of Bordetella pertussis adenylate cyclase toxin. J. Biol. Chem. 2006, 281, 16914–16926. [Google Scholar]
- Guermonprez, P.; Khelef, N.; Blouin, E.; Rieu, P.; Ricciardi-Castagnoli, P.; Guiso, N.; Ladant, D.; Leclerc, C. The adenylate cyclase toxin of Bordetella pertussis binds to target cells via the alpha(M)beta(2) integrin (CD11b/CD18). J. Exp. Med. 2001, 193, 1035–1044. [Google Scholar] [CrossRef]
- Carbonetti, N.H. Pertussis toxin and adenylate cyclase toxin: Key virulence factors of Bordetella pertussis and cell biology tools. Future Microbiol. 2010, 5, 455–469. [Google Scholar] [CrossRef]
- Basler, M.; Masin, J.; Osicka, R.; Sebo, P. Pore-forming and enzymatic activities of Bordetella pertussis adenylate cyclase toxin synergize in promoting lysis of monocytes. Infect. Immun. 2006, 74, 2207–2214. [Google Scholar]
- Boyd, A.P.; Ross, P.J.; Conroy, H.; Mahon, N.; Lavelle, E.C.; Mills, K.H. Bordetella pertussis adenylate cyclase toxin modulates innate and adaptive immune responses: Distinct roles for acylation and enzymatic activity in immunomodulation and cell death. J. Immunol. 2005, 175, 730–738. [Google Scholar]
- Hritonenko, V.; Mun, J.J.; Tam, C.; Simon, N.C.; Barbieri, J.T.; Evans, D.J.; Fleiszig, S.M. Adenylate cyclase activity of Pseudomonas aeruginosa ExoY can mediate bleb-niche formation in epithelial cells and contributes to virulence. Microb. Pathog. 2011, 51, 305–312. [Google Scholar] [CrossRef]
- Mock, M.; Fouet, A. Anthrax. Annu. Rev. Microbiol. 2001, 55, 647–671. [Google Scholar] [CrossRef]
- Mourez, M.; Lacy, D.B.; Cunningham, K.; Legmann, R.; Sellman, B.R.; Mogridge, J.; Collier, R.J. 2001: A year of major advances in anthrax toxin research. Trends Microbiol. 2002, 10, 287–293. [Google Scholar] [CrossRef]
- Oncu, S.; Oncu, S.; Sakarya, S. Anthrax—An overview. Med. Sci. Monit. 2003, 9, RA276–283. [Google Scholar]
- Hicks, C.W.; Sweeney, D.A.; Cui, X.; Li, Y.; Eichacker, P.Q. An overview of anthrax infection including the recently identified form of disease in injection drug users. Intensive Care Med. 2012, 38, 1092–1104. [Google Scholar] [CrossRef]
- Sweeney, D.A.; Hicks, C.W.; Cui, X.; Li, Y.; Eichacker, P.Q. Anthrax infection. Am. J. Respir. Crit. Care Med. 2011, 184, 1333–1341. [Google Scholar] [CrossRef]
- Young, J.A.; Collier, R.J. Anthrax toxin: Receptor binding, internalization, pore formation, and translocation. Annu. Rev. Biochem. 2007, 76, 243–265. [Google Scholar] [CrossRef]
- Guichard, A.; Nizet, V.; Bier, E. New insights into the biological effects of anthrax toxins: Linking cellular to organismal responses. Microbes Infect. 2012, 14, 97–118. [Google Scholar] [CrossRef]
- Bann, J.G. Anthrax toxin protective antigen - insights into molecular switching from prepore to pore. Protein Sci. 2012, 21, 1–12. [Google Scholar] [CrossRef]
- Feld, G.K.; Kintzer, A.F.; Tang, I.I.; Thoren, K.L.; Krantz, B.A. Domain flexibility modulates the heterogeneous assembly mechanism of anthrax toxin protective antigen. J. Mol. Biol. 2012, 415, 159–174. [Google Scholar] [CrossRef]
- Feld, G.K.; Thoren, K.L.; Kintzer, A.F.; Sterling, H.J.; Tang, I.I.; Greenberg, S.G.; Williams, E.R.; Krantz, B.A. Structural basis for the unfolding of anthrax lethal factor by protective antigen oligomers. Nat. Struct. Mol. Biol. 2010, 17, 1383–1390. [Google Scholar]
- Christensen, K.A.; Krantz, B.A.; Melnyk, R.A.; Collier, R.J. Interaction of the 20 kDa and 63 kDa fragments of anthrax protective antigen: Kinetics and thermodynamics. Biochemistry 2005, 44, 1047–1053. [Google Scholar]
- Kintzer, A.F.; Sterling, H.J.; Tang, I.I.; Abdul-Gader, A.; Miles, A.J.; Wallace, B.A.; Williams, E.R.; Krantz, B.A. Role of the protective antigen octamer in the molecular mechanism of anthrax lethal toxin stabilization in plasma. J. Mol. Biol. 2010, 399, 741–758. [Google Scholar] [CrossRef]
- Kintzer, A.F.; Sterling, H.J.; Tang, I.I.; Williams, E.R.; Krantz, B.A. Anthrax toxin receptor drives protective antigen oligomerization and stabilizes the heptameric and octameric oligomer by a similar mechanism. PLoS One 2010, 5, e13888. [Google Scholar]
- Kintzer, A.F.; Thoren, K.L.; Sterling, H.J.; Dong, K.C.; Feld, G.K.; Tang, I.I.; Zhang, T.T.; Williams, E.R.; Berger, J.M.; Krantz, B.A. The protective antigen component of anthrax toxin forms functional octameric complexes. J. Mol. Biol. 2009, 392, 614–629. [Google Scholar] [CrossRef]
- Salles, II; Voth, D.E.; Ward, S.C.; Averette, K.M.; Tweten, R.K.; Bradley, K.A.; Ballard, J.D. Cytotoxic activity of Bacillus anthracis protective antigen observed in a macrophage cell line overexpressing ANTXR1. Cell. Microbiol. 2006, 8, 1272–1281. [Google Scholar] [CrossRef]
- Agrawal, A.; Lingappa, J.; Leppla, S.H.; Agrawal, S.; Jabbar, A.; Quinn, C.; Pulendran, B. Impairment of dendritic cells and adaptive immunity by anthrax lethal toxin. Nature 2003, 424, 329–334. [Google Scholar]
- Park, J.M.; Greten, F.R.; Li, Z.W.; Karin, M. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science 2002, 297, 2048–2051. [Google Scholar]
- Xu, L.; Frucht, D.M. Bacillus anthracis: A multi-faceted role for anthrax lethal toxin in thwarting host immune defenses. Int. J. Biochem. Cell Biol. 2007, 39, 20–24. [Google Scholar] [CrossRef]
- Young, J.J.; Bromberg-White, J.L.; Zylstra, C.; Church, J.T.; Boguslawski, E.; Resau, J.H.; Williams, B.O.; Duesbery, N.S. LRP5 and LRP6 are not required for protective antigen-mediated internalization or lethality of anthrax lethal toxin. PLoS Pathog. 2007, 3, e27. [Google Scholar] [CrossRef]
- Ali, S.R.; Timmer, A.M.; Bilgrami, S.; Park, E.J.; Eckmann, L.; Nizet, V.; Karin, M. Anthrax toxin induces macrophage death by p38 MAPK inhibition but leads to inflammasome activation via ATP leakage. Immunity 2011, 35, 34–44. [Google Scholar] [CrossRef]
- Cao, S.; Guo, A.; Wu, G.; Liu, Z.; Chen, W.; Feng, C.; Zhang, C.C.; Chen, H. Residue histidine 669 is essential for the catalytic activity of Bacillus anthracis lethal factor. J. Bacteriol. 2010, 192, 5799–5805. [Google Scholar] [CrossRef]
- Chow, E.M.; Batty, S.; Mogridge, J. Anthrax lethal toxin promotes dephosphorylation of TTP and formation of processing bodies. Cell. Microbiol. 2010, 12, 557–568. [Google Scholar] [CrossRef]
- Dalkas, G.A.; Chasapis, C.T.; Gkazonis, P.V.; Bentrop, D.; Spyroulias, G.A. Conformational dynamics of the anthrax lethal factor catalytic center. Biochemistry 2010, 49, 10767–10769. [Google Scholar]
- Dumas, E.K.; Cox, P.M.; Fullenwider, C.O.; Nguyen, M.; Centola, M.; Frank, M.B.; Dozmorov, I.; James, J.A.; Farris, A.D. Anthrax lethal toxin-induced gene expression changes in mouse lung. Toxins 2011, 3, 1111–1130. [Google Scholar] [CrossRef]
- Ebrahimi, C.M.; Sheen, T.R.; Renken, C.W.; Gottlieb, R.A.; Doran, K.S. Contribution of lethal toxin and edema toxin to the pathogenesis of anthrax meningitis. Infect. Immun. 2011, 79, 2510–2518. [Google Scholar] [CrossRef]
- Lee, S.; Wang, Y.; Kim, S.O.; Han, J. AMPD3 is involved in anthrax LeTx-induced macrophage cell death. Protein Cell 2011, 2, 564–572. [Google Scholar] [CrossRef]
- Liu, T.; Milia, E.; Warburton, R.R.; Hill, N.S.; Gaestel, M.; Kayyali, U.S. Anthrax lethal toxin disrupts the endothelial permeability barrier through blocking p38 signaling. J. Cell. Physiol. 2012, 227, 1438–1445. [Google Scholar] [CrossRef]
- Sun, C.; Fang, H.; Xie, T.; Auth, R.D.; Patel, N.; Murray, P.R.; Snoy, P.J.; Frucht, D.M. Anthrax lethal toxin disrupts intestinal barrier function and causes systemic infections with enteric bacteria. PLoS One 2012, 7, e33583. [Google Scholar]
- Tamayo, A.G.; Slater, L.; Taylor-Parker, J.; Bharti, A.; Harrison, R.; Hung, D.T.; Murphy, J.R. GRP78(BiP) facilitates the cytosolic delivery of anthrax lethal factor (LF) in vivo and functions as an unfoldase in vitro. Mol. Microbiol. 2011, 81, 1390–1401. [Google Scholar]
- Thomas, J.; Epshtein, Y.; Chopra, A.; Ordog, B.; Ghassemi, M.; Christman, J.W.; Nattel, S.; Cook, J.L.; Levitan, I. Anthrax lethal factor activates K(+) channels to induce IL-1beta secretion in macrophages. J. Immunol. 2011, 186, 5236–5243. [Google Scholar]
- Vuyisich, M.; Sanders, C.K.; Graves, S.W. Binding and cell intoxication studies of anthrax lethal toxin. Mol. Biol. Rep. 2012, 39, 5897–5903. [Google Scholar] [CrossRef]
- Xie, T.; Auth, R.D.; Frucht, D.M. The effects of anthrax lethal toxin on host barrier function. Toxins 2011, 3, 591–607. [Google Scholar] [CrossRef]
- Pellizzari, R.; Guidi-Rontani, C.; Vitale, G.; Mock, M.; Montecucco, C. Anthrax lethal factor cleaves MKK3 in macrophages and inhibits the LPS/IFNgamma-induced release of NO and TNFalpha. FEBS Lett. 1999, 462, 199–204. [Google Scholar] [CrossRef]
- Vitale, G.; Bernardi, L.; Napolitani, G.; Mock, M.; Montecucco, C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem. J. 2000, 352 Pt 3, 739–745. [Google Scholar]
- Vitale, G.; Pellizzari, R.; Recchi, C.; Napolitani, G.; Mock, M.; Montecucco, C. Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem. Biophys. Res. Commun. 1998, 248, 706–711. [Google Scholar] [CrossRef]
- Vitale, G.; Pellizzari, R.; Recchi, C.; Napolitani, G.; Mock, M.; Montecucco, C. Anthrax lethal factor cleaves the N-terminus of MAPKKS and induces tyrosine/threonine phosphorylation of MAPKS in cultured macrophages. J. Appl. Microbiol. 1999, 87, 288. [Google Scholar]
- Ascenzi, P.; Visca, P.; Ippolito, G.; Spallarossa, A.; Bolognesi, M.; Montecucco, C. Anthrax toxin: A tripartite lethal combination. FEBS Lett. 2002, 531, 384–388. [Google Scholar] [CrossRef]
- Brossier, F.; Mock, M. Toxins of Bacillus anthracis. Toxicon 2001, 39, 1747–1755. [Google Scholar] [CrossRef]
- Brossier, F.; Weber-Levy, M.; Mock, M.; Sirard, J.C. Role of toxin functional domains in anthrax pathogenesis. Infect. Immun. 2000, 68, 1781–1786. [Google Scholar] [CrossRef]
- Cunningham, K.; Lacy, D.B.; Mogridge, J.; Collier, R.J. Mapping the lethal factor and edema factor binding sites on oligomeric anthrax protective antigen. Proc. Natl. Acad. Sci. USA 2002, 99, 7049–7053. [Google Scholar]
- Dal Molin, F.; Tonello, F.; Ladant, D.; Zornetta, I.; Zamparo, I.; Di Benedetto, G.; Zaccolo, M.; Montecucco, C. Cell entry and cAMP imaging of anthrax edema toxin. EMBO J. 2006, 25, 5405–5413. [Google Scholar] [CrossRef]
- Gnade, B.T.; Moen, S.T.; Chopra, A.K.; Peterson, J.W.; Yeager, L.A. Emergence of anthrax edema toxin as a master manipulator of macrophage and B cell functions. Toxins 2010, 2, 1881–1897. [Google Scholar] [CrossRef]
- Larabee, J.L.; Maldonado-Arocho, F.J.; Pacheco, S.; France, B.; DeGiusti, K.; Shakir, S.M.; Bradley, K.A.; Ballard, J.D. Glycogen synthase kinase 3 activation is important for anthrax edema toxin-induced dendritic cell maturation and anthrax toxin receptor 2 expression in macrophages. Infect. Immun. 2011, 79, 3302–3308. [Google Scholar]
- Szarowicz, S.E.; During, R.L.; Li, W.; Quinn, C.P.; Tang, W.J.; Southwick, F.S. Bacillus anthracis edema toxin impairs neutrophil actin-based motility. Infect. Immun. 2009, 77, 2455–2464. [Google Scholar] [CrossRef]
- Yeager, L.A.; Chopra, A.K.; Peterson, J.W. Bacillus anthracis edema toxin suppresses human macrophage phagocytosis and cytoskeletal remodeling via the protein kinase A and exchange protein activated by cyclic AMP pathways. Infect. Immun. 2009, 77, 2530–2543. [Google Scholar]
- Thoren, K.L.; Krantz, B.A. The unfolding story of anthrax toxin translocation. Mol. Microbiol. 2011, 80, 588–595. [Google Scholar] [CrossRef]
- Bradley, K.A.; Mogridge, J.; Mourez, M.; Collier, R.J.; Young, J.A. Identification of the cellular receptor for anthrax toxin. Nature 2001, 414, 225–229. [Google Scholar]
- Hong, J.; Beeler, J.; Zhukovskaya, N.L.; He, W.; Tang, W.J.; Rosner, M.R. Anthrax edema factor potency depends on mode of cell entry. Biochem. Biophys. Res. Commun. 2005, 335, 850–857. [Google Scholar] [CrossRef]
- Hong, J.; Doebele, R.C.; Lingen, M.W.; Quilliam, L.A.; Tang, W.J.; Rosner, M.R. Anthrax edema toxin inhibits endothelial cell chemotaxis via Epac and Rap1. J. Biol. Chem. 2007, 282, 19781–19787. [Google Scholar]
- Liu, S.; Leppla, S.H. Cell surface tumor endothelium marker 8 cytoplasmic tail-independent anthrax toxin binding, proteolytic processing, oligomer formation, and internalization. J. Biol. Chem. 2003, 278, 5227–5234. [Google Scholar]
- Santelli, E.; Bankston, L.A.; Leppla, S.H.; Liddington, R.C. Crystal structure of a complex between anthrax toxin and its host cell receptor. Nature 2004, 430, 905–908. [Google Scholar]
- Scobie, H.M.; Marlett, J.M.; Rainey, G.J.; Lacy, D.B.; Collier, R.J.; Young, J.A. Anthrax toxin receptor 2 determinants that dictate the pH threshold of toxin pore formation. PLoS One 2007, 2, e329. [Google Scholar]
- Scobie, H.M.; Rainey, G.J.; Bradley, K.A.; Young, J.A. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 5170–5174. [Google Scholar]
- Bell, S.E.; Mavila, A.; Salazar, R.; Bayless, K.J.; Kanagala, S.; Maxwell, S.A.; Davis, G.E. Differential gene expression during capillary morphogenesis in 3D collagen matrices: Regulated expression of genes involved in basement membrane matrix assembly, cell cycle progression, cellular differentiation and G-protein signaling. J. Cell Sci. 2001, 114, 2755–2773. [Google Scholar]
- Carson-Walter, E.B.; Watkins, D.N.; Nanda, A.; Vogelstein, B.; Kinzler, K.W.; St Croix, B. Cell surface tumor endothelial markers are conserved in mice and humans. Cancer Res. 2001, 61, 6649–6655. [Google Scholar]
- Deuquet, J.; Lausch, E.; Superti-Furga, A.; van der Goot, F.G. The dark sides of capillary morphogenesis gene 2. EMBO J 2012, 31, 3–13. [Google Scholar]
- Fu, S.; Tong, X.; Cai, C.; Zhao, Y.; Wu, Y.; Li, Y.; Xu, J.; Zhang, X.C.; Xu, L.; Chen, W.; et al. The structure of tumor endothelial marker 8 (TEM8) extracellular domain and implications for its receptor function for recognizing anthrax toxin. PLoS One 2010, 5, e11203. [Google Scholar]
- Klimpel, K.R.; Molloy, S.S.; Thomas, G.; Leppla, S.H. Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proc. Natl. Acad. Sci. USA 1992, 89, 10277–10281. [Google Scholar]
- Milne, J.C.; Furlong, D.; Hanna, P.C.; Wall, J.S.; Collier, R.J. Anthrax protective antigen forms oligomers during intoxication of mammalian cells. J. Biol. Chem. 1994, 269, 20607–20612. [Google Scholar]
- Petosa, C.; Collier, R.J.; Klimpel, K.R.; Leppla, S.H.; Liddington, R.C. Crystal structure of the anthrax toxin protective antigen. Nature 1997, 385, 833–838. [Google Scholar]
- Abrami, L.; Liu, S.; Cosson, P.; Leppla, S.H.; van der Goot, F.G. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J. Cell Biol. 2003, 160, 321–328. [Google Scholar] [CrossRef]
- Christensen, K.A.; Krantz, B.A.; Collier, R.J. Assembly and disassembly kinetics of anthrax toxin complexes. Biochemistry 2006, 45, 2380–2386. [Google Scholar]
- Gordon, V.M.; Leppla, S.H.; Hewlett, E.L. Inhibitors of receptor-mediated endocytosis block the entry of Bacillus anthracis adenylate cyclase toxin but not that of Bordetella pertussis adenylate cyclase toxin. Infect. Immun. 1988, 56, 1066–1069. [Google Scholar]
- Mogridge, J.; Cunningham, K.; Lacy, D.B.; Mourez, M.; Collier, R.J. The lethal and edema factors of anthrax toxin bind only to oligomeric forms of the protective antigen. Proc. Natl. Acad. Sci. USA 2002, 99, 7045–7048. [Google Scholar]
- Mogridge, J.; Cunningham, K.; Collier, R.J. Stoichiometry of anthrax toxin complexes. Biochemistry 2002, 41, 1079–1082. [Google Scholar]
- Benson, E.L.; Huynh, P.D.; Finkelstein, A.; Collier, R.J. Identification of residues lining the anthrax protective antigen channel. Biochemistry 1998, 37, 3941–3948. [Google Scholar]
- Friedlander, A.M. Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J. Biol. Chem. 1986, 261, 7123–7126. [Google Scholar]
- Paccani, S.R.; Baldari, C.T. T cell targeting by anthrax toxins: Two faces of the same coin. Toxins 2011, 3, 660–671. [Google Scholar] [CrossRef]
- Janowiak, B.E.; Jennings-Antipov, L.D.; Collier, R.J. Cys-Cys cross-linking shows contact between the N-terminus of lethal factor and Phe427 of the anthrax toxin pore. Biochemistry 2011, 50, 3512–3516. [Google Scholar] [CrossRef]
- Katayama, H.; Janowiak, B.E.; Brzozowski, M.; Juryck, J.; Falke, S.; Gogol, E.P.; Collier, R.J.; Fisher, M.T. GroEL as a molecular scaffold for structural analysis of the anthrax toxin pore. Nat. Struct. Mol. Biol. 2008, 15, 754–760. [Google Scholar]
- Katayama, H.; Wang, J.; Tama, F.; Chollet, L.; Gogol, E.P.; Collier, R.J.; Fisher, M.T. Three-dimensional structure of the anthrax toxin pore inserted into lipid nanodiscs and lipid vesicles. Proc. Natl. Acad. Sci. USA 2010, 107, 3453–3457. [Google Scholar]
- Pilpa, R.M.; Bayrhuber, M.; Marlett, J.M.; Riek, R.; Young, J.A. A receptor-based switch that regulates anthrax toxin pore formation. PLoS Pathog. 2011, 7, e1002354. [Google Scholar] [CrossRef]
- Brown, M.J.; Thoren, K.L.; Krantz, B.A. Charge requirements for proton gradient-driven translocation of anthrax toxin. J. Biol. Chem. 2011, 286, 23189–23199. [Google Scholar]
- Basilio, D.; Kienker, P.K.; Briggs, S.W.; Finkelstein, A. A kinetic analysis of protein transport through the anthrax toxin channel. J. Gen. Physiol. 2011, 137, 521–531. [Google Scholar] [CrossRef]
- Duesbery, N.S.; Webb, C.P.; Leppla, S.H.; Gordon, V.M.; Klimpel, K.R.; Copeland, T.D.; Ahn, N.G.; Oskarsson, M.K.; Fukasawa, K.; Paull, K.D.; et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 1998, 280, 734–737. [Google Scholar]
- Baldari, C.T.; Tonello, F.; Paccani, S.R.; Montecucco, C. Anthrax toxins: A paradigm of bacterial immune suppression. Trends Immunol. 2006, 27, 434–440. [Google Scholar] [CrossRef]
- Rossi Paccani, S.; Tonello, F.; Patrussi, L.; Capitani, N.; Simonato, M.; Montecucco, C.; Baldari, C.T. Anthrax toxins inhibit immune cell chemotaxis by perturbing chemokine receptor signalling. Cell. Microbiol. 2007, 9, 924–929. [Google Scholar] [CrossRef]
- Kirby, J.E. Anthrax lethal toxin induces human endothelial cell apoptosis. Infect. Immun. 2004, 72, 430–439. [Google Scholar] [CrossRef]
- Laine, E.; Martinez, L.; Blondel, A.; Malliavin, T.E. Activation of the edema factor of Bacillus anthracis by calmodulin: Evidence of an interplay between the EF-calmodulin interaction and calcium binding. Biophys. J 2010, 99, 2264–2272. [Google Scholar] [CrossRef]
- Dal Molin, F.; Zornetta, I.; Puhar, A.; Tonello, F.; Zaccolo, M.; Montecucco, C. cAMP imaging of cells treated with pertussis toxin, cholera toxin, and anthrax edema toxin. Biochem. Biophys. Res. Commun. 2008, 376, 429–433. [Google Scholar] [CrossRef]
- Pezard, C.; Berche, P.; Mock, M. Contribution of individual toxin components to virulence of Bacillus anthracis. Infect. Immun. 1991, 59, 3472–3477. [Google Scholar]
- Stanley, J.L.; Smith, H. Purification of factor 1 and recognition of a third factor of the anthrax toxin. J. Gen. Microbiol. 1961, 26, 49–63. [Google Scholar] [CrossRef]
- Lovchik, J.A.; Drysdale, M.; Koehler, T.M.; Hutt, J.A.; Lyons, C.R. Expression of either Lethal Toxin or Edema Toxin by Bacillus anthracis is Sufficient for Virulence in a Rabbit Model of Inhalational Anthrax. Infect. Immun. 2012. [Google Scholar] [CrossRef]
- Confer, D.L.; Eaton, J.W. Phagocyte impotence caused by an invasive bacterial adenylate cyclase. Science 1982, 217, 948–950. [Google Scholar]
- Leppla, S.H. Anthrax toxin edema factor: A bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. USA 1982, 79, 3162–3166. [Google Scholar] [CrossRef]
- Leppla, S.H. Purification and characterization of adenylyl cyclase from Bacillus anthracis. Methods Enzymol. 1991, 195, 153–168. [Google Scholar]
- O´Brien, J.; Friedlander, A.; Dreier, T.; Ezzell, J.; Leppla, S. Effects of anthrax toxin components on human neutrophils. Infect. Immun. 1985, 47, 306–310. [Google Scholar]
- Tournier, J.N.; Quesnel-Hellmann, A.; Mathieu, J.; Montecucco, C.; Tang, W.J.; Mock, M.; Vidal, D.R.; Goossens, P.L. Anthrax edema toxin cooperates with lethal toxin to impair cytokine secretion during infection of dendritic cells. J. Immunol. 2005, 174, 4934–4941. [Google Scholar]
- Crawford, M.A.; Aylott, C.V.; Bourdeau, R.W.; Bokoch, G.M. Bacillus anthracis toxins inhibit human neutrophil NADPH oxidase activity. J. Immunol. 2006, 176, 7557–7565. [Google Scholar]
- Larabee, J.L.; Shakir, S.M.; Hightower, L.; Ballard, J.D. Adenomatous polyposis coli protein associates with C/EBP beta and increases Bacillus anthracis edema toxin-stimulated gene expression in macrophages. J. Biol. Chem. 2011, 286, 19364–19372. [Google Scholar]
- Voth, D.E.; Hamm, E.E.; Nguyen, L.G.; Tucker, A.E.; Salles, I.I.; Ortiz-Leduc, W.; Ballard, J.D. Bacillus anthracis oedema toxin as a cause of tissue necrosis and cell type-specific cytotoxicity. Cell. Microbiol. 2005, 7, 1139–1149. [Google Scholar] [CrossRef]
- Paccani, S.R.; Tonello, F.; Ghittoni, R.; Natale, M.; Muraro, L.; D´Elios, M.M.; Tang, W.J.; Montecucco, C.; Baldari, C.T. Anthrax toxins suppress T lymphocyte activation by disrupting antigen receptor signaling. J. Exp. Med. 2005, 201, 325–331. [Google Scholar] [CrossRef]
- Puhar, A.; Dal Molin, F.; Horvath, S.; Ladant, D.; Montecucco, C. Anthrax edema toxin modulates PKA- and CREB-dependent signaling in two phases. PLoS One 2008, 3, e3564. [Google Scholar]
- Firoved, A.M.; Moayeri, M.; Wiggins, J.F.; Shen, Y.; Tang, W.J.; Leppla, S.H. Anthrax edema toxin sensitizes DBA/2J mice to lethal toxin. Infect. Immun. 2007, 75, 2120–2125. [Google Scholar]
- Hicks, C.W.; Cui, X.; Sweeney, D.A.; Li, Y.; Barochia, A.; Eichacker, P.Q. The potential contributions of lethal and edema toxins to the pathogenesis of anthrax associated shock. Toxins 2011, 3, 1185–1202. [Google Scholar] [CrossRef]
- Dumetz, F.; Jouvion, G.; Khun, H.; Glomski, I.J.; Corre, J.P.; Rougeaux, C.; Tang, W.J.; Mock, M.; Huerre, M.; Goossens, P.L. Noninvasive imaging technologies reveal edema toxin as a key virulence factor in anthrax. Am. J. Pathol. 2011, 178, 2523–2535. [Google Scholar] [CrossRef]
- Brey, R.N. Molecular basis for improved anthrax vaccines. Adv. Drug Deliv. Rev. 2005, 57, 1266–1292. [Google Scholar] [CrossRef]
- Scorpio, A.; Blank, T.E.; Day, W.A.; Chabot, D.J. Anthrax vaccines: Pasteur to the present. Cell. Mol. Life Sci. 2006, 63, 2237–2248. [Google Scholar] [CrossRef]
- Scorpio, A.; Chabot, D.J.; Day, W.A.; O´Brien, D.K.; Vietri, N.J.; Itoh, Y.; Mohamadzadeh, M.; Friedlander, A.M. Poly-γ-glutamate capsule-degrading enzyme treatment enhances phagocytosis and killing of encapsulated Bacillus anthracis. Antimicrob. Agents. Chemother. 2007, 51, 215–222. [Google Scholar]
- Tournier, J.N.; Ulrich, R.G.; Quesnel-Hellmann, A.; Mohamadzadeh, M.; Stiles, B.G. Anthrax, toxins and vaccines: A 125-year journey targeting Bacillus anthracis. Expert Rev. Anti Infect. Ther. 2009, 7, 219–236. [Google Scholar] [CrossRef]
- Riddle, V.; Leese, P.; Blanset, D.; Adamcio, M.; Meldorf, M.; Lowy, I. Phase I study evaluating the safety and pharmacokinetics of MDX-1303, a fully human monoclonal antibody against Bacillus anthracis protective antigen, in healthy volunteer. Clin. Vaccine Immunol. 2011, 18, 2136–2142. [Google Scholar] [CrossRef]
- Little, S.F.; Webster, W.M.; Fisher, D.E. Monoclonal antibodies directed against protective antigen of Bacillus anthracis enhance lethal toxin activity in vivo. FEMS Immunol. Med. Microbiol. 2011, 62, 11–22. [Google Scholar] [CrossRef]
- vor dem Esche, U.; Huber, M.; Zgaga-Griesz, A.; Grunow, R.; Beyer, W.; Hahn, U.; Bessler, W.G. Passive vaccination with a human monoclonal antibody: Generation of antibodies and studies for efficacy in Bacillus anthracis infections. Immunobiology 2011, 216, 847–853. [Google Scholar] [CrossRef]
- Chitlaru, T.; Altboum, Z.; Reuveny, S.; Shafferman, A. Progress and novel strategies in vaccine development and treatment of anthrax. Immunol. Rev. 2011, 239, 221–236. [Google Scholar] [CrossRef]
- Altaweel, L.; Chen, Z.; Moayeri, M.; Cui, X.; Li, Y.; Su, J.; Fitz, Y.; Johnson, S.; Leppla, S.H.; Purcell, R.; et al. Delayed treatment with W1-mAb, a chimpanzee-derived monoclonal antibody against protective antigen, reduces mortality from challenges with anthrax edema or lethal toxin in rats and with anthrax spores in mice. Crit. Care Med. 2011, 39, 1439–1447. [Google Scholar]
- Makiya, M.; Dolan, M.; Agulto, L.; Purcell, R.; Chen, Z. Structural basis of anthrax edema factor neutralization by a neutralizing antibody. Biochem. Biophys. Res. Commun. 2012, 417, 324–329. [Google Scholar] [CrossRef]
- Leysath, C.E.; Chen, K.H.; Moayeri, M.; Crown, D.; Fattah, R.; Chen, Z.; Das, S.R.; Purcell, R.H.; Leppla, S.H. Mouse monoclonal antibodies to anthrax edema factor protect against infection. Infect. Immun. 2011, 79, 4609–4616. [Google Scholar] [CrossRef]
- Chen, Z.; Moayeri, M.; Purcell, R. Monoclonal Antibody Therapies against Anthrax. Toxins 2011, 3, 1004–1019. [Google Scholar] [CrossRef]
- Crowe, S.R.; Garman, L.; Engler, R.J.; Farris, A.D.; Ballard, J.D.; Harley, J.B.; James, J.A. Anthrax vaccination induced anti-lethal factor IgG: Fine specificity and neutralizing capacity. Vaccine 2011, 29, 3670–3678. [Google Scholar]
- Kulshreshtha, P.; Bhatnagar, R. Inhibition of anthrax toxins with a bispecific monoclonal antibody that cross reacts with edema factor as well as lethal factor of Bacillus anthracis. Mol. Immunol. 2011, 48, 1958–1965. [Google Scholar] [CrossRef]
- Pini, A.; Runci, Y.; Falciani, C.; Lelli, B.; Brunetti, J.; Pileri, S.; Fabbrini, M.; Lozzi, L.; Ricci, C.; Bernini, A. et al. Stable peptide inhibitors prevent binding of lethal and oedema factors to protective antigen and neutralize anthrax toxin in vivo. Biochem. J. 2006, 395, 157–163. [Google Scholar] [CrossRef]
- Laine, E.; Goncalves, C.; Karst, J.C.; Lesnard, A.; Rault, S.; Tang, W.J.; Malliavin, T.E.; Ladant, D.; Blondel, A. Use of allostery to identify inhibitors of calmodulin-induced activation of Bacillus anthracis edema factor. Proc. Natl. Acad. Sci. USA 2010, 107, 11277–11282. [Google Scholar]
- Dessauer, C.W.; Tesmer, J.J.; Sprang, S.R.; Gilman, A.G. The interactions of adenylate cyclases with P-site inhibitors. Trends Pharmacol. Sci. 1999, 20, 205–210. [Google Scholar] [CrossRef]
- Tesmer, J.J.; Dessauer, C.W.; Sunahara, R.K.; Murray, L.D.; Johnson, R.A.; Gilman, A.G.; Sprang, S.R. Molecular basis for P-site inhibition of adenylyl cyclase. Biochemistry 2000, 39, 14464–14471. [Google Scholar]
- Johnson, R.A.; Shoshani, I. Inhibition of Bordetella pertussis and Bacillus anthracis adenylyl cyclases by polyadenylate and "P"-site agonists. J. Biol. Chem. 1990, 265, 19035–19039. [Google Scholar]
- Shen, Y.; Zhukovskaya, N.L.; Zimmer, M.I.; Soelaiman, S.; Bergson, P.; Wang, C.R.; Gibbs, C.S.; Tang, W.J. Selective inhibition of anthrax edema factor by adefovir, a drug for chronic hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 3242–3247. [Google Scholar]
- Soelaiman, S.; Wei, B.Q.; Bergson, P.; Lee, Y.S.; Shen, Y.; Mrksich, M.; Shoichet, B.K.; Tang, W.J. Structure-based inhibitor discovery against adenylyl cyclase toxins from pathogenic bacteria that cause anthrax and whooping cough. J. Biol. Chem. 2003, 278, 25990–25997. [Google Scholar]
- Lee, Y.S.; Bergson, P.; He, W.S.; Mrksich, M.; Tang, W.J. Discovery of a small molecule that inhibits the interaction of anthrax edema factor with its cellular activator, calmodulin. Chem. Biol. 2004, 11, 1139–1146. [Google Scholar] [CrossRef]
- Gille, A.; Seifert, R. MANT-substituted guanine nucleotides: A novel class of potent adenylyl cyclase inhibitors. Life Sci. 2003, 74, 271–279. [Google Scholar] [CrossRef]
- Gille, A.; Seifert, R. 2′(3′)-O-(N-methylanthraniloyl)-substituted GTP analogs: A novel class of potent competitive adenylyl cyclase inhibitors. J. Biol. Chem. 2003, 278, 12672–12679. [Google Scholar] [CrossRef]
- Taha, H.; Dove, S.; Geduhn, J.; König, B.; Shen, Y.; Tang, W.J.; Seifert, R. Inhibition of the adenylyl cyclase toxin, edema factor, from Bacillus anthracis by a series of 18 mono- and bis-(M)ANT-substituted nucleoside 5′-triphosphates. Naunyn Schmiedebergs Arch. Pharmacol. 2012, 385, 57–68. [Google Scholar] [CrossRef]
- Taha, H.M.; Schmidt, J.; Göttle, M.; Suryanarayana, S.; Shen, Y.; Tang, W.J.; Gille, A.; Geduhn, J.; König, B.; Dove, S.; et al. Molecular analysis of the interaction of anthrax adenylyl cyclase toxin, edema factor, with 2′(3′)-O-(N-(methyl)anthraniloyl)-substituted purine and pyrimidine nucleotides. Mol. Pharmacol. 2009, 75, 693–703. [Google Scholar] [CrossRef]
- Gille, A.; Lushington, G.H.; Mou, T.C.; Doughty, M.B.; Johnson, R.A.; Seifert, R. Differential inhibition of adenylyl cyclase isoforms and soluble guanylyl cyclase by purine and pyrimidine nucleotides. J. Biol. Chem. 2004, 279, 19955–19969. [Google Scholar]
- Geduhn, J.; Dove, S.; Shen, Y.; Tang, W.J.; König, B.; Seifert, R. Bis-halogen-anthraniloyl-substituted nucleoside 5′-triphosphates as potent and selective inhibitors of Bordetella pertussis adenylyl cyclase toxin. J. Pharmacol. Exp. Ther. 2011, 336, 104–115. [Google Scholar] [CrossRef]
- Gaion, R.M.; Krishna, G. Cytidylate cyclase: Possible artifacts in the methodology. Science 1979, 203, 672–673. [Google Scholar]
- Gaion, R.M.; Krishna, G. Cytidylate cyclase: The product isolated by the method of Cech and Ignarro is not cytidine 3′,5′-monophosphate. Biochem. Biophys. Res. Commun. 1979, 86, 105–111. [Google Scholar] [CrossRef]
- Göttle, M.; Dove, S.; Kees, F.; Schlossmann, J.; Geduhn, J.; König, B.; Shen, Y.; Tang, W.J.; Kaever, V.; Seifert, R. Cytidylyl and uridylyl cyclase activity of Bacillus anthracis edema factor and Bordetella pertussis CyaA. Biochemistry 2010, 49, 5494–5503. [Google Scholar]
- Flynn, G.E.; Zagotta, W.N. A cysteine scan of the inner vestibule of cyclic nucleotide-gated channels reveals architecture and rearrangement of the pore. J. Gen. Physiol. 2003, 121, 563–582. [Google Scholar] [CrossRef]
- Sunderman, E.R.; Zagotta, W.N. Sequence of events underlying the allosteric transition of rod cyclic nucleotide-gated channels. J. Gen. Physiol. 1999, 113, 621–640. [Google Scholar] [CrossRef]
- Sunderman, E.R.; Zagotta, W.N. Mechanism of allosteric modulation of rod cyclic nucleotide-gated channels. J. Gen. Physiol. 1999, 113, 601–620. [Google Scholar] [CrossRef]
- Postea, O.; Biel, M. Exploring HCN channels as novel drug targets. Nat. Rev. Drug Discov. 2011, 10, 903–914. [Google Scholar]
- Bush, L.M.; Abrams, B.H.; Beall, A.; Johnson, C.C. Index case of fatal inhalational anthrax due to bioterrorism in the United States. N. Engl. J. Med. 2001, 345, 1607–1610. [Google Scholar] [CrossRef]
- Jernigan, J.A.; Stephens, D.S.; Ashford, D.A.; Omenaca, C.; Topiel, M.S.; Galbraith, M.; Tapper, M.; Fisk, T.L.; Zaki, S.; Popovic, T.; et al. Bioterrorism-related inhalational anthrax: The first 10 cases reported in the United States. Emerg. Infect. Dis. 2001, 7, 933–944. [Google Scholar]
- Guarner, J.; Jernigan, J.A.; Shieh, W.J.; Tatti, K.; Flannagan, L.M.; Stephens, D.S.; Popovic, T.; Ashford, D.A.; Perkins, B.A.; Zaki, S.R. Pathology and pathogenesis of bioterrorism-related inhalational anthrax. Am. J. Pathol. 2003, 163, 701–709. [Google Scholar] [CrossRef]
- Nguyen, C.; Feng, C.; Zhan, M.; Cross, A.S.; Goldblum, S.E. Bacillus anthracis-derived edema toxin (ET) counter-regulates movement of neutrophils and macromolecules through the endothelial paracellular pathway. BMC Microbiol. 2012, 12, 2. [Google Scholar] [CrossRef]
- Twenhafel, N.A.; Leffel, E.; Pitt, M.L. Pathology of inhalational anthrax infection in the african green monkey. Vet. Pathol. 2007, 44, 716–721. [Google Scholar] [CrossRef]
- He, P.; Zeng, M.; Curry, F.E. Dominant role of cAMP in regulation of microvessel permeability. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1124–1133. [Google Scholar]
- Waschke, J.; Drenckhahn, D.; Adamson, R.H.; Barth, H.; Curry, F.E. cAMP protects endothelial barrier functions by preventing Rac-1 inhibition. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2427–2433. [Google Scholar] [CrossRef]
- Fukuhara, S.; Sakurai, A.; Sano, H.; Yamagishi, A.; Somekawa, S.; Takakura, N.; Saito, Y.; Kangawa, K.; Mochizuki, N. Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol. Cell. Biol. 2005, 25, 136–146. [Google Scholar]
- Drum, C.L.; Yan, S.Z.; Bard, J.; Shen, Y.Q.; Lu, D.; Soelaiman, S.; Grabarek, Z.; Bohm, A.; Tang, W.J. Structural basis for the activation of anthrax adenylyl cyclase exotoxin by calmodulin. Nature 2002, 415, 396–402. [Google Scholar]
- Shen, Y.; Zhukovskaya, N.L.; Guo, Q.; Florian, J.; Tang, W.J. Calcium-independent calmodulin binding and two-metal-ion catalytic mechanism of anthrax edema factor. EMBO J 2005, 24, 929–941. [Google Scholar] [CrossRef]
- Guo, Q.; Shen, Y.; Lee, Y.S.; Gibbs, C.S.; Mrksich, M.; Tang, W.J. Structural basis for the interaction of Bordetella pertussis adenylyl cyclase toxin with calmodulin. EMBO J. 2005, 24, 3190–3201. [Google Scholar] [CrossRef]
- Zea, C.J.; Camci-Unal, G.; Pohl, N.L. Thermodynamics of binding of divalent magnesium and manganese to uridine phosphates: Implications for diabetes-related hypomagnesaemia and carbohydrate biocatalysis. Chem. Cent. J. 2008, 2, 15. [Google Scholar] [CrossRef]
- Bock, C.W.; Kaufman Katz, A.; Markham, G.D.; Glusker, J.P. Manganese as a Replacement for Magnesium and Zinc: Functional Comparison of the Divalent Ions. J. Am. Chem. Soc. 1999, 121, 7360–7372. [Google Scholar]
- Newton, R.P.; Salih, S.G.; Salvage, B.J.; Kingston, E.E. Extraction, purification and identification of cytidine 3′,5′-cyclic monophosphate from rat tissues. Biochem. J. 1984, 221, 665–673. [Google Scholar]
- Newton, R.P.; Salvage, B.J.; Hakeem, N.A. Cytidylate cyclase: Development of assay and determination of kinetic properties of a cytidine 3′,5′-cyclic monophosphate-synthesizing enzyme. Biochem. J. 1990, 265, 581–586. [Google Scholar]
- Newton, R.P.; Groot, N.; van Geyschem, J.; Diffley, P.E.; Walton, T.J.; Bayliss, M.A.; Harris, F.M.; Games, D.E.; Brenton, A.G. Estimation of cytidylyl cyclase activity and monitoring of side-product formation by fast-atom bombardment mass spectrometry. Rapid Commun. Mass Spectrom. 1997, 11, 189–194. [Google Scholar]
- Newton, R.P.; Evans, A.M.; van Geyschem, J.; Diffley, P.J.; Hassam, H.G.; Hakeem, N.A.; Moyse, C.D.; Cooke, R.; Salvage, B.J. Radioimmunoassay of cytidine 3′,5′-cyclic monophosphate: Unambiguous assay by means of an optimized protocol incorporating a trilayer column separation to obviate cross-reactivity problems. J. Immunoassay 1994, 15, 317–337. [Google Scholar] [CrossRef]
- Newton, R.P.; Kingston, E.E.; Hakeem, N.A.; Salih, S.G.; Beynon, J.H.; Moyse, C.D. Extraction, purification, identification and metabolism of 3′,5′-cyclic UMP, 3′,5′-cyclic IMP and 3′,5′-cyclic dTMP from rat tissues. Biochem. J. 1986, 236, 431–439. [Google Scholar]
- Elliott, G.R.; Lauwen, A.P.; Bonta, I.L. Dibutyryl cytidine 3′:5′-cyclic monophosphate; an inhibitor of A23187-stimulated macrophage leukotriene B4 synthesis. Agents Actions 1991, 32, 90–91. [Google Scholar] [CrossRef]
- Ervens, J.; Seifert, R. Differential modulation by N4,2′-O-dibutyryl cytidine 3′:5′-cyclic monophosphate of neutrophil activation. Biochem. Biophys. Res. Commun. 1991, 174, 258–267. [Google Scholar] [CrossRef]
- Burhenne, H.; Beste, K.Y.; Spangler, C.M.; Voigt, U.; Kaever, V.; Seifert, R. Determination of cytidine 3′,5′-cyclic monophosphate and uridine 3′,5′-cyclic monophosphate in mammalian cell systems and in human urine by highperformance liquid chromatography/mass spectrometry. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2011, 383 (Suppl. 1), P096. [Google Scholar]
- Taylor, S.S.; Kim, C.; Cheng, C.Y.; Brown, S.H.; Wu, J.; Kannan, N. Signaling through cAMP and cAMP-dependent protein kinase: Diverse strategies for drug design. Biochim. Biophys. Acta 2008, 1784, 16–26. [Google Scholar]
- Kim, C.; Cheng, C.Y.; Saldanha, S.A.; Taylor, S.S. PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell 2007, 130, 1032–1043. [Google Scholar] [CrossRef]
- Taylor, S.S.; Kim, C.; Vigil, D.; Haste, N.M.; Yang, J.; Wu, J.; Anand, G.S. Dynamics of signaling by PKA. Biochim. Biophys. Acta 2005, 1754, 25–37. [Google Scholar]
- Casteel, D.E.; Smith-Nguyen, E.V.; Sankaran, B.; Roh, S.H.; Pilz, R.B.; Kim, C. A crystal structure of the cyclic GMP-dependent protein kinase I{beta} dimerization/docking domain reveals molecular details of isoform-specific anchoring. J. Biol. Chem. 2010, 285, 32684–32688. [Google Scholar]
- Desch, M.; Schinner, E.; Kees, F.; Hofmann, F.; Seifert, R.; Schlossmann, J. Cyclic cytidine 3′,5′-monophosphate (cCMP) signals via cGMP kinase I. FEBS Lett. 2010, 584, 3979–3984. [Google Scholar] [CrossRef]
- Wolter, S.; Golombek, M.; Seifert, R. Differential activation of cAMP- and cGMP-dependent protein kinases by cyclic purine and pyrimidine nucleotides. Biochem. Biophys. Res. Commun. 2011, 415, 563–566. [Google Scholar] [CrossRef]
- Newton, R.P.; Khan, J.A.; Brenton, A.G.; Langridge, J.I.; Harris, F.M.; Walton, T.J. Quantitation by fast-atom bombardment mass spectrometry: Assay of cytidine 3′,5′-cyclic monophosphate-responsive protein kinase. Rapid Commun. Mass Spectrom. 1992, 6, 601–607. [Google Scholar]
- Bond, A.E.; Dudley, E.; Tuytten, R.; Lemiere, F.; Smith, C.J.; Esmans, E.L.; Newton, R.P. Mass spectrometric identification of Rab23 phosphorylation as a response to challenge by cytidine 3′,5′-cyclic monophosphate in mouse brain. Rapid Commun. Mass Spectrom. 2007, 21, 2685–2692. [Google Scholar]
- Ding, S.; Bond, A.E.; Lemiere, F.; Tuytten, R.; Esmans, E.L.; Brenton, A.G.; Dudley, E.; Newton, R.P. Online immobilized metal affinity chromatography/mass spectrometric analysis of changes elicited by cCMP in the murine brain phosphoproteome. Rapid Commun. Mass Spectrom. 2008, 22, 4129–4138. [Google Scholar]
- Hammerschmidt, A.; Chatterji, B.; Zeiser, J.; Schröder, A.; Genieser, H.G.; Pich, A.; Kaever, V.; Schwede, F.; Wolter, S.; Seifert, R. Binding of regulatory subunits of cyclic AMP-dependent protein kinase to cyclic CMP agarose. PLoS One 2012. accepted for publication, PONE-D-12–13426 10.1371/journal.pone.0039848. [Google Scholar]
- Reinecke, D.; Burhenne, H.; Sandner, P.; Kaever, V.; Seifert, R. Human cyclic nucleotide phosphodiesterases possess a much broader substrate-specificity than previously appreciated. FEBS Lett. 2011, 585, 3259–3262. [Google Scholar] [CrossRef]
- Bender, A.T.; Beavo, J.A. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef]
- Conti, M.; Beavo, J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: Essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 2007, 76, 481–511. [Google Scholar] [CrossRef]
- Helfman, D.M.; Katoh, N.; Kuo, J.F. Purification and properties of cyclic CMP phosphodiesterase. Adv. Cyclic Nucleotide Protein Phosphorylation Res. 1984, 16, 403–416. [Google Scholar]
- Newton, R.P.; Salih, S.G. Cyclic CMP phosphodiesterase: Isolation, specificity and kinetic properties. Int. J. Biochem. 1986, 18, 743–752. [Google Scholar] [CrossRef]
- Newton, R.P.; Bayliss, M.A.; Khan, J.A.; Bastani, A.; Wilkins, A.C.; Games, D.E.; Walton, T.J.; Brenton, A.G.; Harris, F.M. Kinetic analysis of cyclic CMP-specific and multifunctional phosphodiesterases by quantitative positive-ion fast-atom bombardment mass spectrometry. Rapid Commun. Mass Spectrom. 1999, 13, 574–584. [Google Scholar] [CrossRef]
- Biel, M. Cyclic nucleotide-regulated cation channels. J. Biol. Chem. 2009, 284, 9017–9021. [Google Scholar] [CrossRef]
- Kaupp, U.B.; Niidome, T.; Tanabe, T.; Terada, S.; Bonigk, W.; Stuhmer, W.; Cook, N.J.; Kangawa, K.; Matsuo, H.; Hirose, T.; et al. Primary structure and functional expression from complementary DNA of the rod photoreceptor cyclic GMP-gated channel. Nature 1989, 342, 762–766. [Google Scholar]
- Contreras, J.E.; Holmgren, M. Access of quaternary ammonium blockers to the internal pore of cyclic nucleotide-gated channels: Implications for the location of the gate. J. Gen. Physiol. 2006, 127, 481–494. [Google Scholar] [CrossRef]
- Varnum, M.D.; Black, K.D.; Zagotta, W.N. Molecular mechanism for ligand discrimination of cyclic nucleotide-gated channels. Neuron 1995, 15, 619–625. [Google Scholar] [CrossRef]
- Shapiro, M.S.; Zagotta, W.N. Structural basis for ligand selectivity of heteromeric olfactory cyclic nucleotide-gated channels. Biophys. J. 2000, 78, 2307–2320. [Google Scholar]
- Zong, X.; Krause, S.; Chen, C.C.; Gruner, C.; Cao-Ehlker, X.; Fenske, S.; Wahl-Schott, C.; Biel, M. Regulation of HCN channel activity by Cyclic Cytidine 3′,5′-Monophosphate. Naunyn-Schmiedeberg´s Arch. Pharmacol. 2012, 385, S1–S116. [Google Scholar]
- Beste, K.Y.; Burhenne, H.; Kaever, V.; Stasch, J.P.; Seifert, R. Nucleotidyl cyclase activity of soluble guanylyl cyclase alpha1beta1. Biochemistry 2012, 51, 194–204. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Göttle, M.; Dove, S.; Seifert, R. Bacillus anthracis Edema Factor Substrate Specificity: Evidence for New Modes of Action. Toxins 2012, 4, 505-535. https://doi.org/10.3390/toxins4070505
Göttle M, Dove S, Seifert R. Bacillus anthracis Edema Factor Substrate Specificity: Evidence for New Modes of Action. Toxins. 2012; 4(7):505-535. https://doi.org/10.3390/toxins4070505
Chicago/Turabian StyleGöttle, Martin, Stefan Dove, and Roland Seifert. 2012. "Bacillus anthracis Edema Factor Substrate Specificity: Evidence for New Modes of Action" Toxins 4, no. 7: 505-535. https://doi.org/10.3390/toxins4070505
APA StyleGöttle, M., Dove, S., & Seifert, R. (2012). Bacillus anthracis Edema Factor Substrate Specificity: Evidence for New Modes of Action. Toxins, 4(7), 505-535. https://doi.org/10.3390/toxins4070505