Determinants of pH-Dependent Modulation of Translocation in Dermonecrotic G-Protein-Deamidating Toxins

Abstract
:1. Introduction
2. Results and Discussion
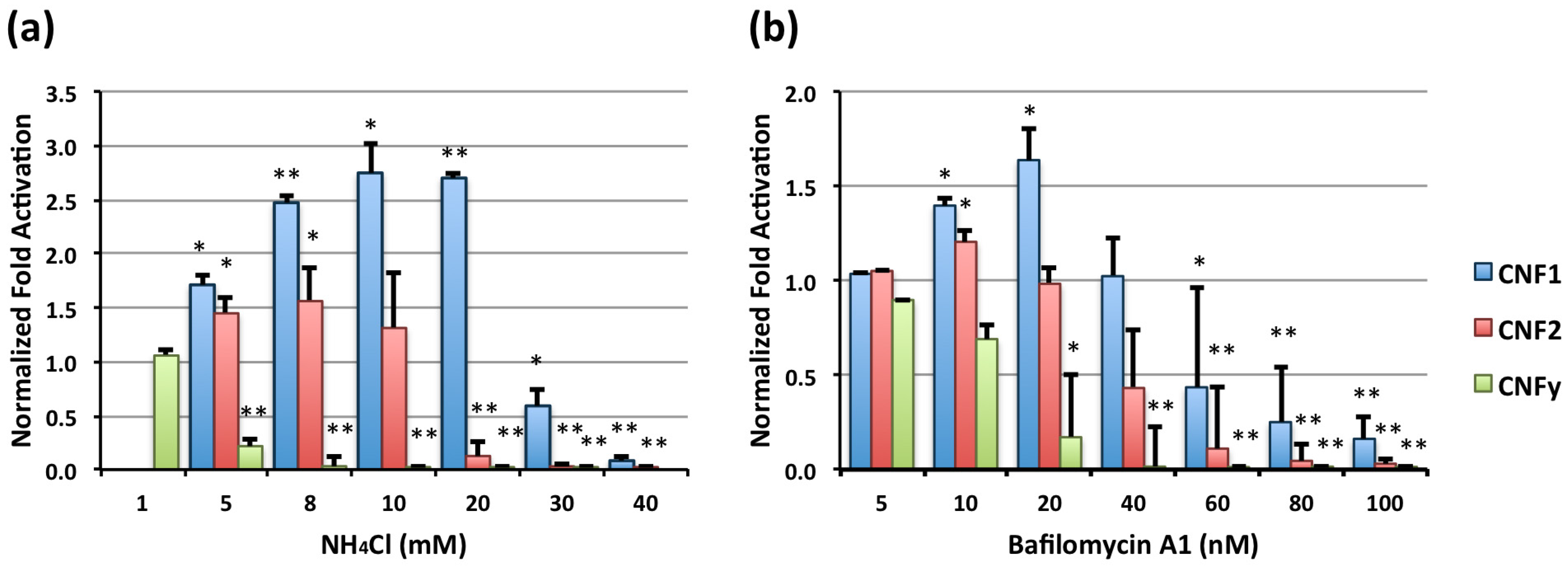
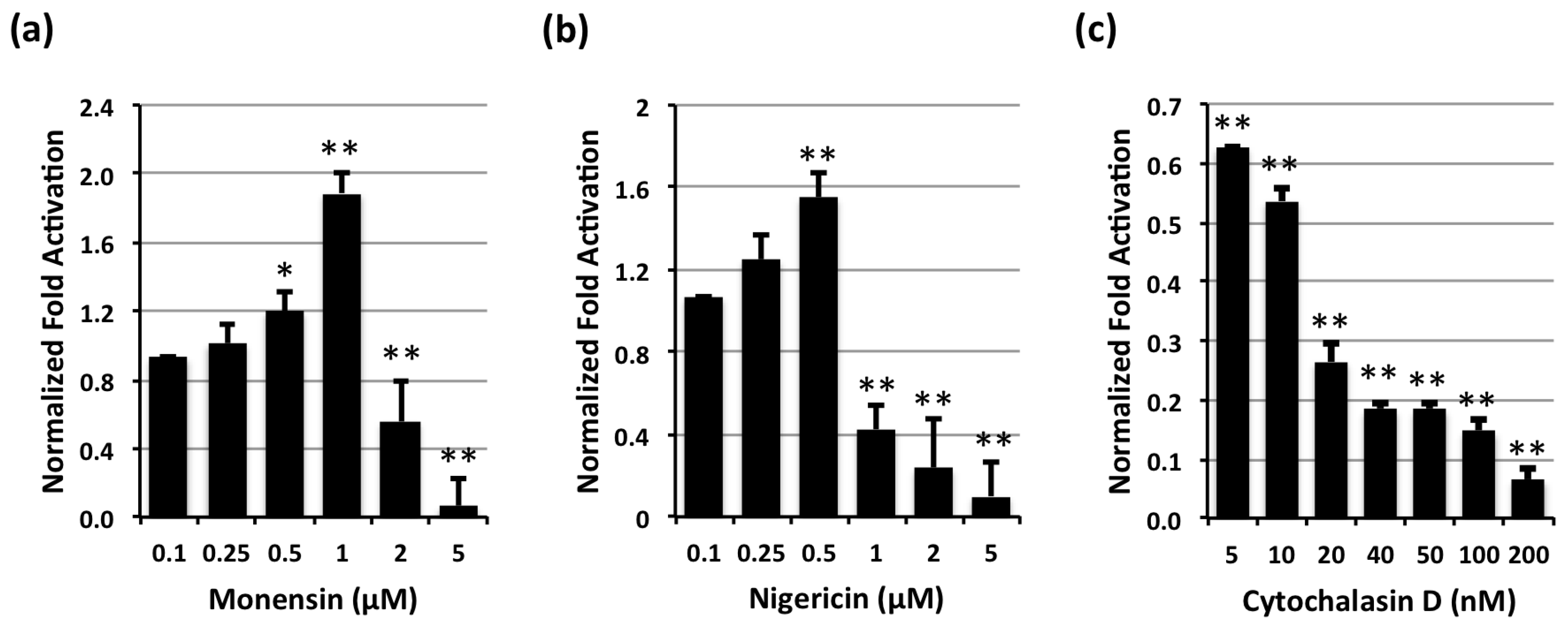
2.1. Effect of Endosomal Acidification Inhibitors on CNF-Mediated SRE-Luciferase Activity


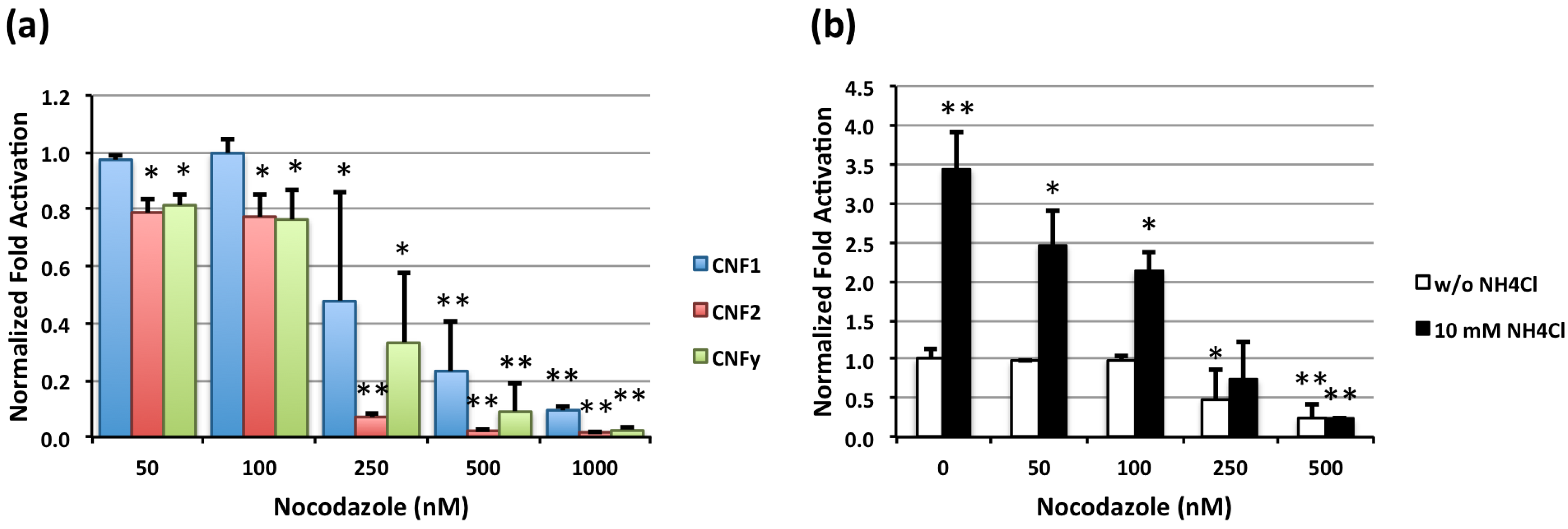
2.2. Effect of Nocodazole on CNF-Mediated SRE-Luciferase Activity and NH4Cl Enhancement of CNF1-Mediated SRE-Luciferase Activity

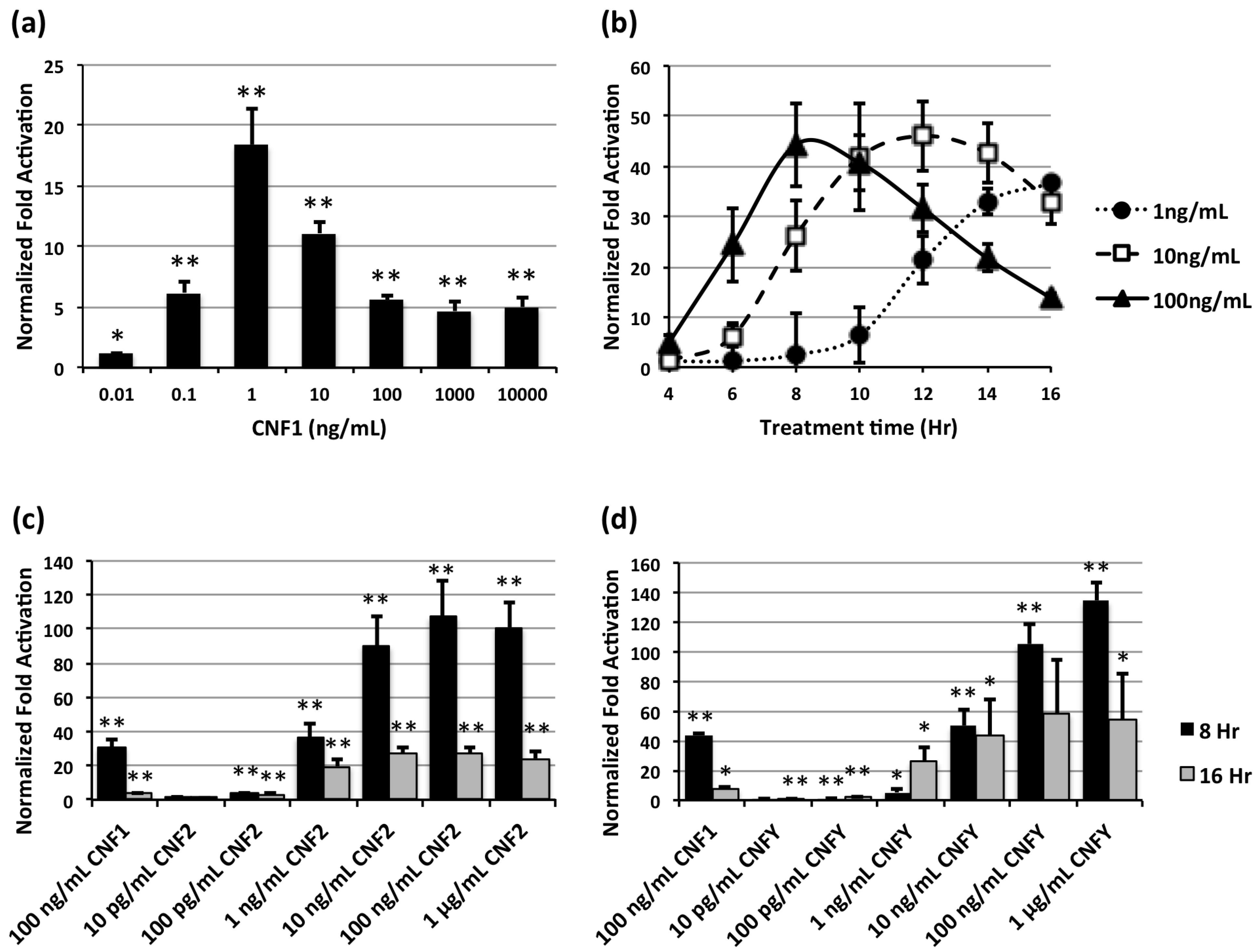
2.3. Comparison of CNF-Mediated SRE-Luciferase Activity

2.4. Comparison of Selected N-Terminal Regions of CNFs and PMT


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Calc. pI value a | Residues 119–267 b | |||||||
|---|---|---|---|---|---|---|---|---|
| 1–536 b | 1–267 | 267–536 | 1–119 | Number R/K | Number D/E | Net Charge | Calc. pI | |
| CNF1 | 5.05 | 6.34 | 4.60 | 4.88 | 21 | 19 | 2 | 8.48 |
| CNF2 | 5.66 | 6.84 | 5.15 | 5.58 | 20 | 19 | 1 | 7.97 |
| CNFy | 4.80 | 5.17 | 4.57 | 5.20 | 18 | 22 | −4 | 5.00 |
| PMT | 4.99 | 5.34 | 4.70 | 5.18 | 16 | 19 | −3 | 5.80 |
3. Experimental Section
3.1. Expression, Purification and Quantification of CNF Proteins
3.2. Cell Culture
3.3. SRE-Luciferase Assay
3.4. Data Analysis
3.5. Treatment of Cells with Toxins and Inhibitors
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Knust, Z.; Schmidt, G. Cytotoxic necrotizing factors (CNFs)—A growing toxin family. Toxins 2010, 2, 116–127. [Google Scholar] [CrossRef]
- Wilson, B.A.; Ho, M. Cellular and molecular action of the mitogenic protein-deamidating toxin from Pasteurella multocida. FEBS J. 2011, 278, 4616–4632. [Google Scholar] [CrossRef]
- Aktories, K. Bacterial protein toxins that modify host regulatory GTPases. Nat. Rev. Microbiol. 2011, 9, 487–498. [Google Scholar] [CrossRef]
- Wilson, B.A.; Ho, M. Recent insights into Pasteurella multocida toxin and other G-protein-modulating bacterial toxins. Future Microbiol. 2010, 5, 1185–1201. [Google Scholar] [CrossRef]
- Chung, J.W.; Hong, S.J.; Kim, K.J.; Goti, D.; Stins, M.F.; Shin, S.; Dawson, V.L.; Dawson, T.M.; Kim, K.S. 37-kDa laminin receptor precursor modulates cytotoxic necrotizing factor 1-mediated RhoA activation and bacterial uptake. J. Biol. Chem. 2003, 278, 16857–16862. [Google Scholar] [CrossRef]
- Kim, K.J.; Chung, J.W.; Kim, K.S. 67-kDa laminin receptor promotes internalization of cytotoxic necrotizing factor 1-expressing Escherichia coli K1 into human brain microvascular endothelial cells. J. Biol. Chem. 2005, 280, 1360–1368. [Google Scholar]
- McNichol, B.A.; Rasmussen, S.B.; Carvalho, H.M.; Meysick, K.C.; O’Brien, A.D. Two domains of cytotoxic necrotizing factor type 1 bind the cellular receptor, laminin receptor precursor protein. Infect. Immun. 2007, 75, 5095–5104. [Google Scholar] [CrossRef]
- Blumenthal, B.; Hoffmann, C.; Aktories, K.; Backert, S.; Schmidt, G. The cytotoxic necrotizing factors from Yersinia pseudotuberculosis and from Escherichia coli bind to different cellular receptors but take the same route to the cytosol. Infect. Immun. 2007, 75, 3344–3353. [Google Scholar] [CrossRef]
- Brothers, M.C.; Ho, M.; Maharjan, R.; Clemons, N.C.; Bannai, Y.; Waites, M.A.; Faulkner, M.J.; Kuhlenschmidt, T.B.; Kuhlenschmidt, M.S.; Blanke, S.R.; et al. Membrane interaction of Pasteurella multocida toxin involves sphingomyelin. FEBS J. 2011, 278, 4633–4648. [Google Scholar] [CrossRef]
- Hinton, A.; Bond, S.; Forgac, M. V-ATPase functions in normal and disease processes. Pflugers Archiv. Eur. J. Physiol. 2009, 457, 589–598. [Google Scholar] [CrossRef]
- Contamin, S.; Galmiche, A.; Doye, A.; Flatau, G.; Benmerah, A.; Boquet, P. The p21 Rho-activating toxin cytotoxic necrotizing factor 1 is endocytosed by a clathrin-independent mechanism and enters the cytosol by an acidic-dependent membrane translocation step. Mol. Biol. Cell 2000, 11, 1775–1787. [Google Scholar]
- Oswald, E.; Sugai, M.; Labigne, A.; Wu, H.C.; Fiorentini, C.; Boquet, P.; O’Brien, A.D. Cytotoxic necrotizing factor type 2 produced by virulent Escherichia coli modifies the small GTP-binding proteins Rho involved in assembly of actin stress fibers. Proc. Natl. Acad. Sci. USA 1994, 91, 3814–3818. [Google Scholar] [CrossRef]
- Baldwin, M.R.; Lakey, J.H.; Lax, A.J. Identification and characterization of the Pasteurella multocida toxin translocation domain. Mol. Microbiol. 2004, 54, 239–250. [Google Scholar] [CrossRef]
- Repella, T.L.; Ho, M.; Chong, T.P.; Bannai, Y.; Wilson, B.A. Arf6-dependent intracellular trafficking of Pasteurella multocida toxin and pH-dependent translocation from late endosomes. Toxins 2011, 3, 218–241. [Google Scholar] [CrossRef]
- Rozengurt, E.; Higgins, T.; Chanter, N.; Lax, A.J.; Staddon, J.M. Pasteurella multocida toxin: Potent mitogen for cultured fibroblasts. Proc. Natl. Acad. Sci. USA 1990, 87, 123–127. [Google Scholar] [CrossRef]
- Mollenhauer, H.H.; Morre, D.J.; Rowe, L.D. Alteration of intracellular traffic by monensin; mechanism, specificity and relationship to toxicity. Biochim. Biophys. Acta 1990, 1031, 225–246. [Google Scholar] [CrossRef]
- Marnell, M.H.; Stookey, M.; Draper, R.K. Monensin blocks the transport of diphtheria toxin to the cell cytoplasm. J. Cell Biol. 1982, 93, 57–62. [Google Scholar] [CrossRef]
- Cooper, J.A. Effects of cytochalasin and phalloidin on actin. J. Cell Biol. 1987, 105, 1473–1478. [Google Scholar] [CrossRef]
- Cover, T.L.; Vaughn, S.G.; Cao, P.; Blaser, M.J. Potentiation of Helicobacter pylori vacuolating toxin activity by nicotine and other weak bases. J. Infect. Dis. 1992, 166, 1073–1078. [Google Scholar] [CrossRef]
- Cover, T.L.; Reddy, L.Y.; Blaser, M.J. Effects of ATPase inhibitors on the response of HeLa cells to Helicobacter pylori vacuolating toxin. Infect. Immun. 1993, 61, 1427–1431. [Google Scholar]
- Mekada, E.; Uchida, T.; Okada, Y. Methylamine stimulates the action of ricin toxin but inhibits that of diphtheria toxin. J. Biol. Chem. 1981, 256, 1225–1228. [Google Scholar]
- Sandvig, K.; Olsnes, S. Entry of the toxic proteins abrin, modeccin, ricin, and diphtheria toxin into cells. II. Effect of pH, metabolic inhibitors, and ionophores and evidence for toxin penetration from endocytotic vesicles. J. Biol. Chem. 1982, 257, 7504–7513. [Google Scholar]
- Dyve Lingelem, A.B.; Bergan, J.; Sandvig, K. Inhibitors of intravesicular acidification protect against Shiga toxin in a pH-independent manner. Traffic 2012, 13, 443–454. [Google Scholar] [CrossRef]
- Hudson, T.H.; Grillo, F.G. Brefeldin-A enhancement of ricin A-chain immunotoxins and blockade of intact ricin, modeccin, and abrin. J. Biol. Chem. 1991, 266, 18586–18592. [Google Scholar]
- Rapak, A.; Falnes, P.O.; Olsnes, S. Retrograde transport of mutant ricin to the endoplasmic reticulum with subsequent translocation to cytosol. Proc. Natl. Acad. Sci. USA 1997, 94, 3783–3788. [Google Scholar] [CrossRef]
- Yoshida, T.; Chen, C.C.; Zhang, M.S.; Wu, H.C. Disruption of the Golgi apparatus by brefeldin A inhibits the cytotoxicity of ricin, modeccin, and Pseudomonas toxin. Exp. Cell Res. 1991, 192, 389–395. [Google Scholar] [CrossRef]
- Bomsel, M.; Parton, R.; Kuznetsov, S.A.; Schroer, T.A.; Gruenberg, J. Microtubule- and motor-dependent fusion in vitro between apical and basolateral endocytic vesicles from MDCK cells. Cell 1990, 62, 719–731. [Google Scholar]
- Gruenberg, J.; Griffiths, G.; Howell, K.E. Characterization of the early endosome and putative endocytic carrier vesicles in vivo and with an assay of vesicle fusion in vitro. J. Cell Biol. 1989, 108, 1301–1316. [Google Scholar] [CrossRef]
- Breitfeld, P.P.; McKinnon, W.C.; Mostov, K.E. Effect of nocodazole on vesicular traffic to the apical and basolateral surfaces of polarized MDCK cells. J. Cell Biol. 1990, 111, 2365–2373. [Google Scholar] [CrossRef]
- Maples, C.J.; Ruiz, W.G.; Apodaca, G. Both microtubules and actin filaments are required for efficient postendocytotic traffic of the polymeric immunoglobulin receptor in polarized Madin-Darby canine kidney cells. J. Biol. Chem. 1997, 272, 6741–6751. [Google Scholar] [CrossRef]
- Pei, S.; Doye, A.; Boquet, P. Mutation of specific acidic residues of the CNF1 T domain into lysine alters cell membrane translocation of the toxin. Mol. Microbiol. 2001, 41, 1237–1247. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Compute pI/MW Tool, Excerpt from Chapter: Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Clifton, NJ, USA, 2013. [Google Scholar]
- Aminova, L.R.; Luo, S.; Bannai, Y.; Ho, M.; Wilson, B.A. The C3 domain of Pasteurella multocida toxin is the minimal domain responsible for activation of Gq-dependent calcium and mitogenic signaling. Protein Sci. 2008, 17, 945–949. [Google Scholar] [CrossRef]
- Kingston, R.E.; Chen, C.A.; Okayama, H. Calcium phosphate transfection. Curr. Protoc. Cell Biol. 2003. [Google Scholar] [CrossRef]
- Madhumathi, J.; Verma, R.S. Therapeutic targets and recent advances in protein immunotoxins. Curr. Opin. Microbiol. 2012, 15, 300–309. [Google Scholar] [CrossRef]
- Adkins, I.; Holubova, J.; Kosova, M.; Sadilkova, L. Bacteria and their toxins tamed for immunotherapy. Curr. Pharm. Biotechnol. 2012, 13, 1446–1473. [Google Scholar] [CrossRef]
- Pastan, I.; Hassan, R.; Fitzgerald, D.J.; Kreitman, R.J. Immunotoxin therapy of cancer. Nat. Rev. Cancer 2006, 6, 559–565. [Google Scholar] [CrossRef]
- Ho, M.; Chang, L.H.; Pires-Alves, M.; Thyagarajan, B.; Bloom, J.E.; Gu, Z.; Aberle, K.K.; Teymorian, S.A.; Bannai, Y.; Johnson, S.C.; et al. Recombinant botulinum neurotoxin A heavy chain-based delivery vehicles for neuronal cell targeting. Protein Eng. Des. Sel. 2011, 24, 247–253. [Google Scholar] [CrossRef]
- Krautz-Peterson, G.; Zhang, Y.; Chen, K.; Oyler, G.A.; Feng, H.; Shoemaker, C.B. Retargeting Clostridium difficile toxin B to neuronal cells as a potential vehicle for cytosolic delivery of therapeutic biomolecules to treat botulism. J. Toxicol. 2012, 2012, 760142. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Repella, T.L.; Ho, M.; Wilson, B.A. Determinants of pH-Dependent Modulation of Translocation in Dermonecrotic G-Protein-Deamidating Toxins. Toxins 2013, 5, 1167-1179. https://doi.org/10.3390/toxins5061167
Repella TL, Ho M, Wilson BA. Determinants of pH-Dependent Modulation of Translocation in Dermonecrotic G-Protein-Deamidating Toxins. Toxins. 2013; 5(6):1167-1179. https://doi.org/10.3390/toxins5061167
Chicago/Turabian StyleRepella, Tana L., Mengfei Ho, and Brenda A. Wilson. 2013. "Determinants of pH-Dependent Modulation of Translocation in Dermonecrotic G-Protein-Deamidating Toxins" Toxins 5, no. 6: 1167-1179. https://doi.org/10.3390/toxins5061167