Computational Design of Peptide Ligands for Ochratoxin A



Abstract
:1. Introduction
2. Results and Discussion
2.1. Computational Modelling


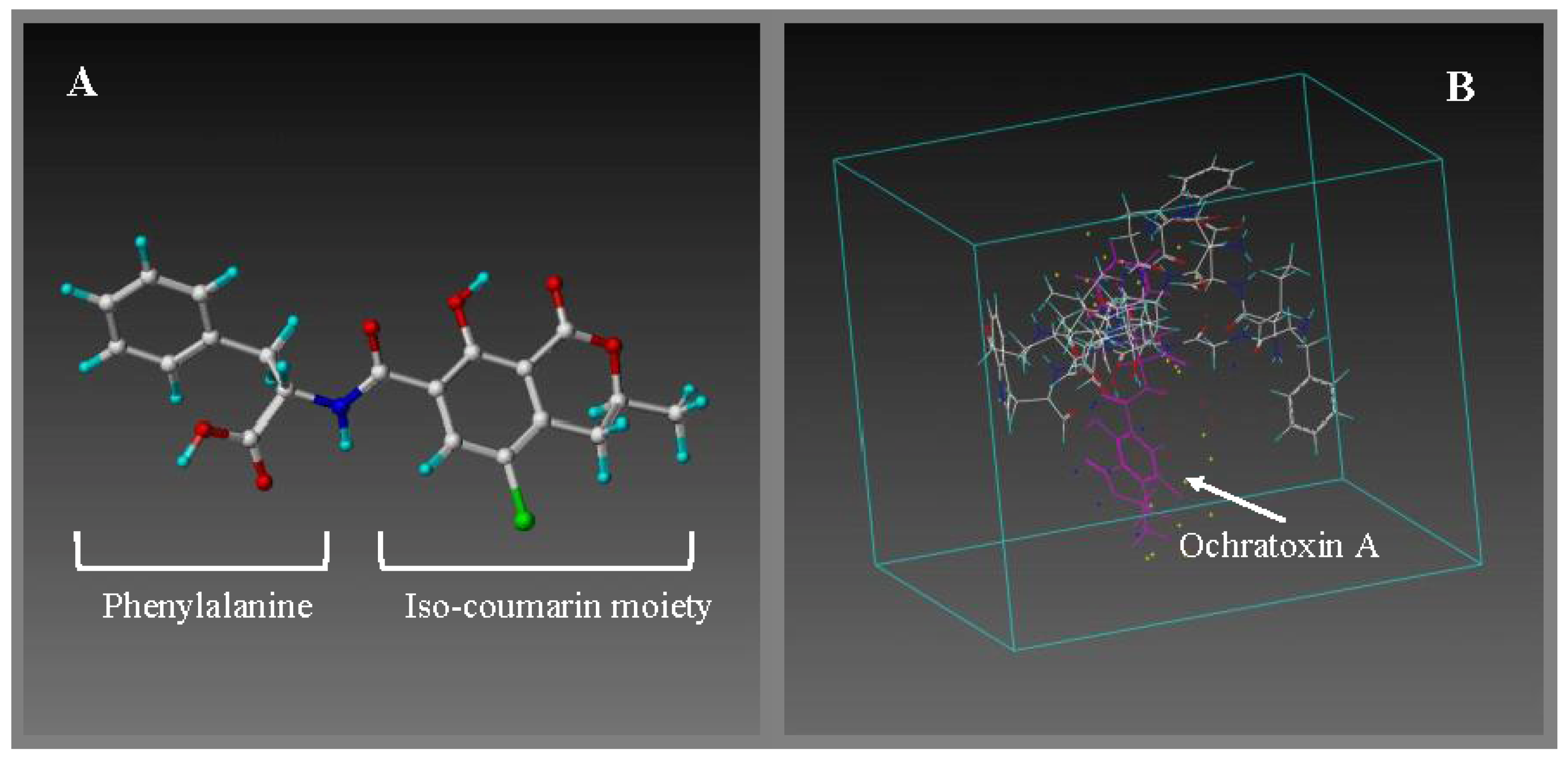{kind=link}
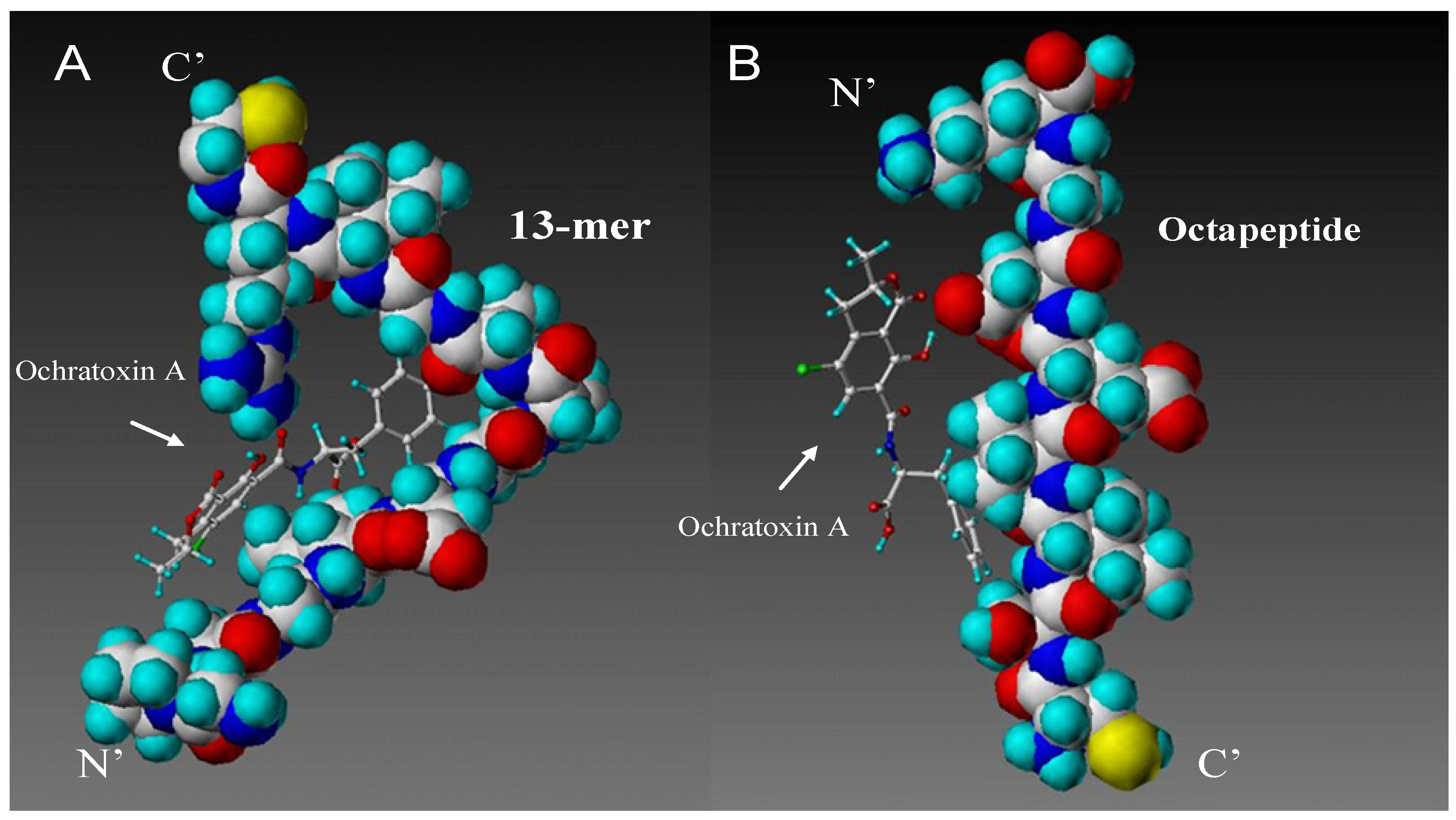{kind=link}
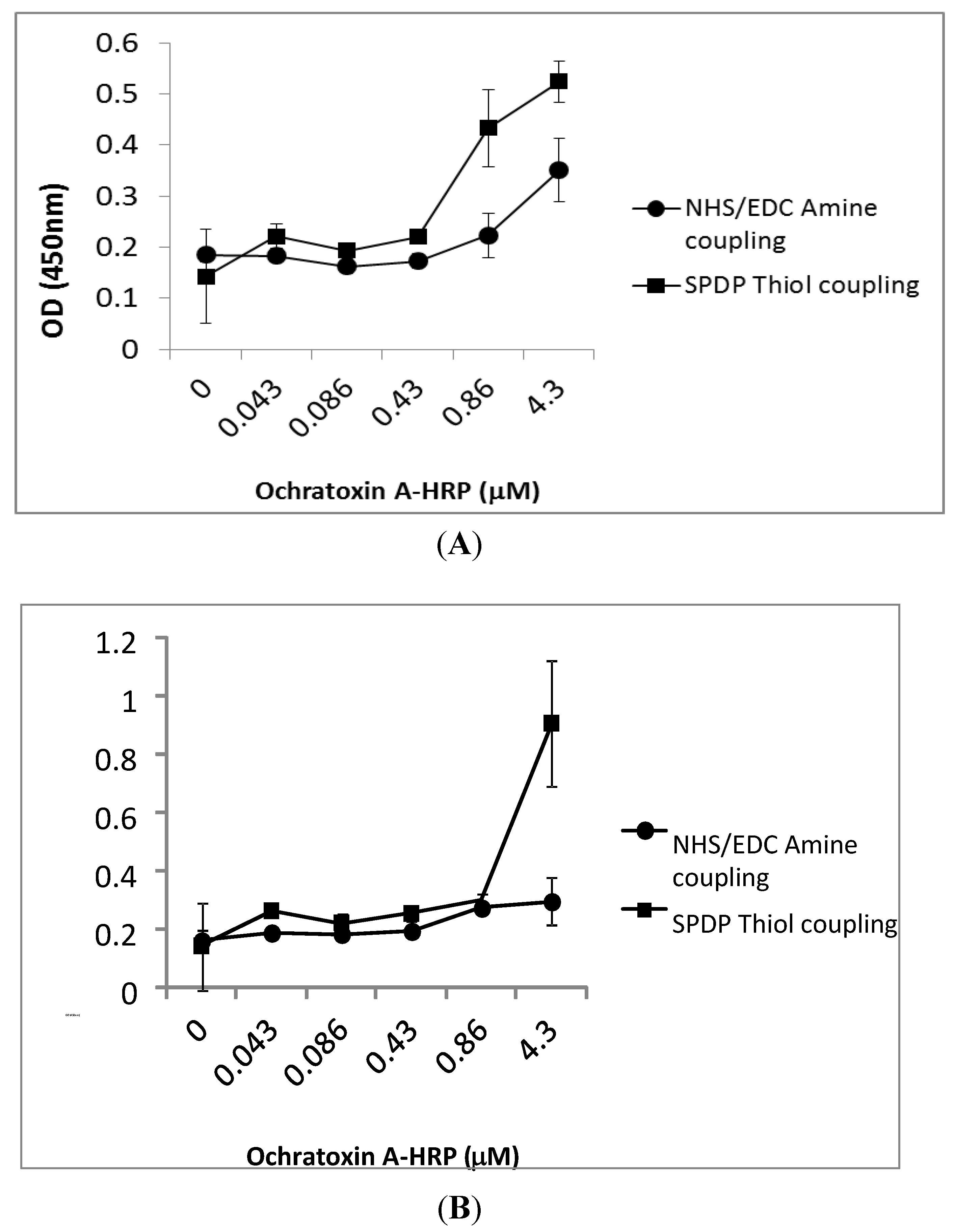{kind=link}
{kind=link}
{kind=link}
| Amino acid | Polarity | Binding energy (kcal mol−1) |
|---|---|---|
| Phenylalanine Phe) | Apolar | −33.52 |
| Proline (Pro) | Apolar | −32.10 |
| Valine (Val) | Apolar | −30.93 |
| Isoleucine (Ile) | Apolar | −30.37 |
| Leucine (Leu) | Apolar | −28.94 |
| Cysteine (Cys) | Polar (uncharged) | −28.67 |
| Tyrosine (Tyr) | Polar (uncharged) | −27.29 |
| Methionine (Met) | Apolar | −26.33 |
| Threonine (Thr) | Polar (uncharged) | −25.55 |
| Tryptophan (Trp) | Apolar | −22.71 |
| Alanine (Ala) | Apolar | −21.87 |
| Glutamate (Glu) | Polar (negatively charged) | −20.63 |
| Aspartate (Asp) | Polar (negatively charged) | −19.86 |
| Asparagine (Asn) | Polar (uncharged) | −13.27 |
| Lysine (Lys) | Polar (positively charged) | −11.72 |
| Histidine (His) | Polar (positively charged) | −10.06 |
| Glutamine (Gln) | Polar (uncharged) | −6.43 |
| Arginine (Arg) | Polar (positively charged) | −5.65 |
| Serine (Ser) | Polar (uncharged) | −5.23 |
| Glycine (Gly) | Polar (uncharged) | −1.89 |
| Sequence | Binding energy (kcal mol−1) |
|---|---|
| Ile-Gly-Ala | −44.55 |
| Ile-Gly-Ala-Pro | −44.54 |
| Ile-Gly-Ala-Gly | −40.05 |
| Ile-Gly-Ala-Cys | −38.56 |
| Ile-Gly-Ala-Pro-Ala | −37.47 |
| Amino acid sequence | Binding energy (kcal mol−1) |
|---|---|
| Pro-Ser-Ile-Val-Glu | −46.45 |
| Ile-Gly-Ala | −44.55 |
| Ile-Gly-Ala-Pro | −44.54 |
| Cys-Ser-Ile-Val-Glu | −42.13 |
| Ile-Gly-Ala-Pro-Ala | −37.47 |
| Cys-Gly-Pro-Ala-Gly-Ile | −31.85 |
| Ser-Pro-Ala-Gly-Ile | −31.56 |
| Peptide sequence | Binding energy (kcal mol−1) |
|---|---|
| Gly-Pro-Ser-Ile-Val-Glu-Cys | −17.24 |
| Pro-Ser-Ile-Val-Glu-Pro-Ser-Ile-Val-Glu-Cys | −16.72 |
| Ser-Pro-Ala-Gly-Ile | −16.07 |
| Cys-Ser-Ile-Val-Glu-Asp-Gly-Lys | −14.90 |
| Cys-Gln-Ile-Val-Glu-Pro-Gln-Ile-Val-Glu | −14.63 |
| Cys-Phe-Asp-Pro-Ala-Gly-Ile-Lys | −14.25 |
| Cys-Phe-Asp-Ala-Pro-Ala-Gly-Ile-Lys | −13.08 |
| Pro-Ser-Ile-Val-Glu | −12.48 |
| Gly-Pro-Ala-Gly-Ile-Asp-Gly-Pro-Ala-Gly-Ile-Arg-Cys | −11.81 |
| Gly-Ser-Pro-Ala-Gly-Ile-Gly | −11.78 |
| Cys-Gly-Pro-Ala-Gly-Ile | −8.72 |

2.2. Binding Assay of Ochratoxin A-HRP to the 13-mer and Octapeptide

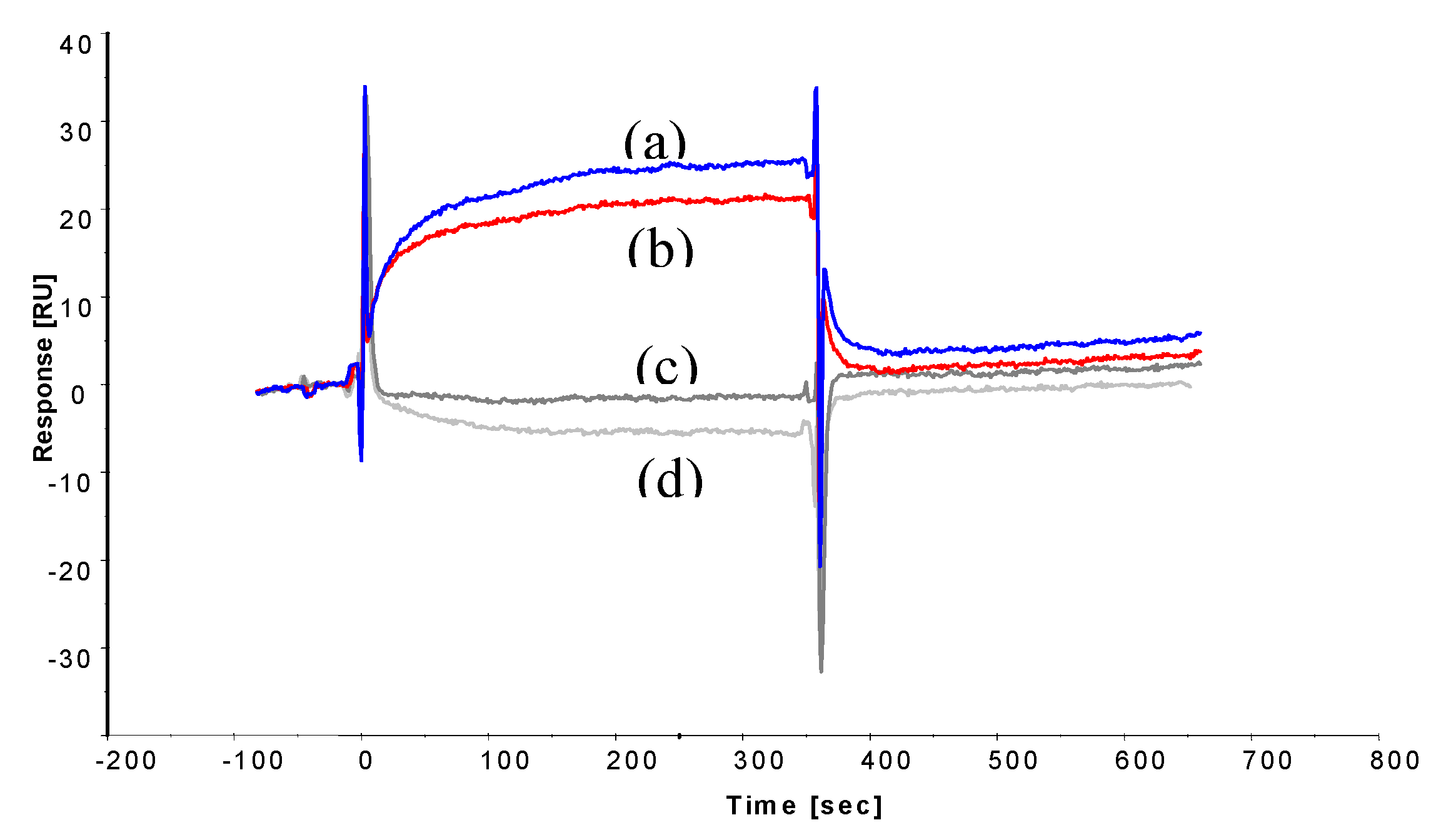
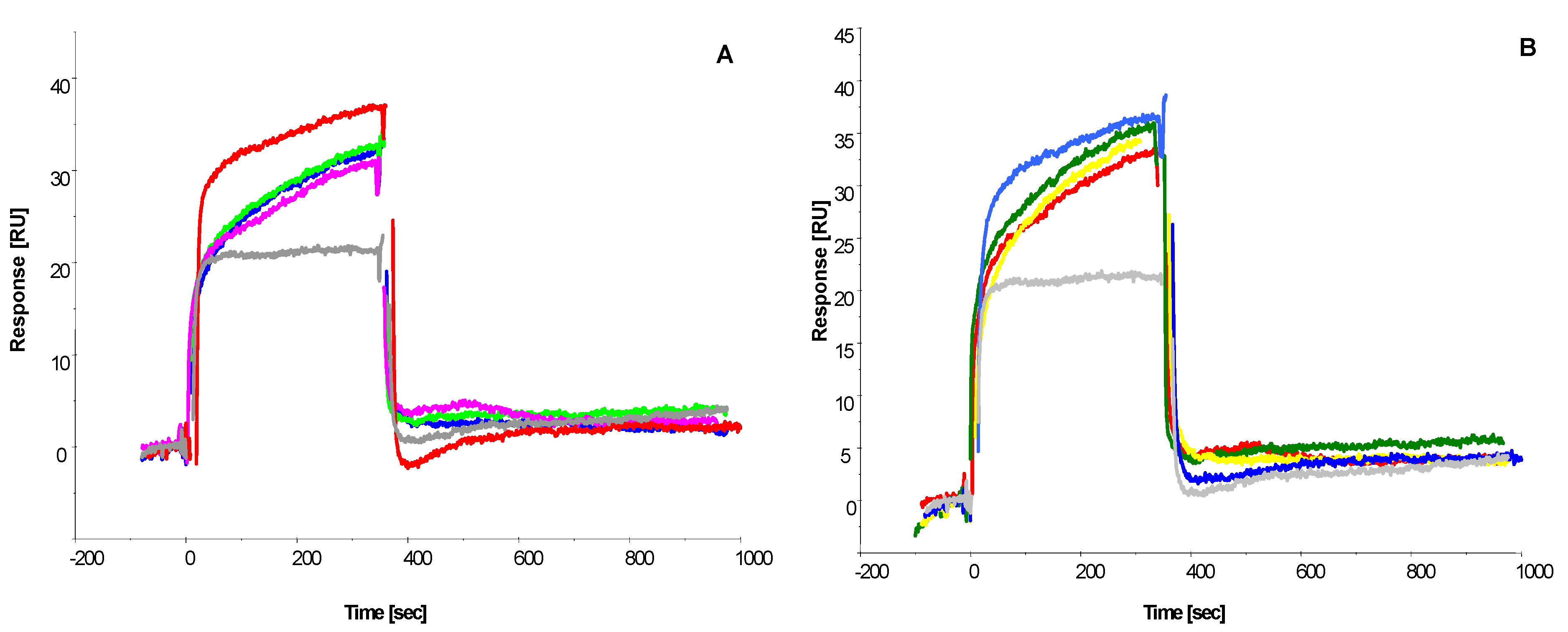
2.3. SPR Analysis to Determine Binding Affinity


3. Experimental Section
3.1. Materials and Reagents
3.2. Computational Modelling
3.3. Solid Phase Binding Assay
3.4. SPR Testing for Binding Interaction
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Adams, M.; Moss, M. Food Microbiology; The Royal Society of Chemistry: Cambridge, UK, 1995; pp. 192–203. [Google Scholar]
- Makun, H.; Gbodi, T.; Akanya, O.; Salako, E.; Ogbadu, G. Fungi and some mycotoxins contaminating rice (Oryza sativa) in Niger state, Nigeria. Afr. J. Biotechnol. 2010, 6, 99–108. [Google Scholar]
- Sulyok, M.; Krska, R.; Schuhmacher, R. Application of an LC-MS/MS based multi-mycotoxin method for the semi-quantitative determination of mycotoxins occurring in different types of food infected by moulds. Food Chem. 2010, 119, 408–416. [Google Scholar] [CrossRef]
- Wild, C.P.; Gong, Y.Y. Mycotoxins and human disease: A largely ignored global health issue. Carcinogenesis 2010, 31, 71–82. [Google Scholar] [CrossRef]
- Bhat, R.; Rai, R.V.; Karim, A. Mycotoxins in food and feed: Present status and future concerns. Compr. Rev. Food Sci. Food Saf. 2010, 9, 57–81. [Google Scholar] [CrossRef]
- Bhatnagar, D.; Brown, R.; Ehrlich, K.; Cleveland, T.E. Mycotoxins contaminating cereal grain crops: Their occurrence and toxicity. Appl. Mycol. Biotechnol. 2002, 2, 171–196. [Google Scholar] [CrossRef]
- Adjou, E.S.; Dahouenon-Ahoussi, E.; Soumanou, M.M. Investigations on the mycoflora and processing effects on the nutritional quality of peanut (Arachis hypogea L. var. TS 32-1). J. Microbiol. Biotechnol. Food Sci. 2012, 2, 1025–1039. [Google Scholar]
- Storari, M.; Dennert, F.G.; Bigler, L.; Gessler, C.; Broggini, G.A. Isolation of mycotoxins producing black aspergilli in herbal teas available on the Swiss market. Food Control 2012, 26, 157–161. [Google Scholar]
- Borbély, M.; Sipos, P.; Pelles, F.; Győri, Z. Mycotoxin contamination in cereals. J. Agronom. Proc. Technol. 2010, 16, 96–98. [Google Scholar]
- Rolle-Kampczyk, U.; Müller, A.; Diez, U.; Rehwagen, M.; Schwenke, A.; Metzner, G.; Herbarth, O. Mycotoxins in house dust—An underestimated problem? Mycotoxin Res. 2000, 16, 100–104. [Google Scholar] [CrossRef]
- Hibi, D.; Suzuki, Y.; Ishii, Y.; Jin, M.; Watanabe, M.; Sugita-Konishi, Y.; Yanai, T.; Nohmi, T.; Nishikawa, A.; Umemura, T. Site-specific in vivo mutagenicity in the kidney of gpt delta rats given a carcinogenic dose of ochratoxin A. Toxicol. Sci. 2011, 122, 406–414. [Google Scholar] [CrossRef]
- Stoev, S.D. Studies on carcinogenic and toxic effects of ochratoxin A in chicks. Toxins 2010, 2, 649–664. [Google Scholar] [CrossRef]
- Czakai, K.; Müller, K.; Mosesso, P.; Pepe, G.; Schulze, M.; Gohla, A.; Patnaik, D.; Dekant, W.; Higgins, J.M.; Mally, A. Perturbation of mitosis through inhibition of histone acetyltransferases: The key to ochratoxin a toxicity and carcinogenicity? Toxicol. Sci. 2011, 122, 317–329. [Google Scholar] [CrossRef]
- Reddy, L.; Bhoola, K. Ochratoxins—Food contaminants: Impact on human health. Toxins 2010, 2, 771–779. [Google Scholar] [CrossRef]
- Pfohl-Leszkowicz, A.; Manderville, R. Review on Ochratoxin A: An overview on toxicity and carcinogenicity in animals and humans. Mol. Nutr. Food Res. 2007, 51, 61–99. [Google Scholar] [CrossRef]
- Pfohl-Leszkowicz, A.; Manderville, R.A. An update on direct genotoxicity as molecular mechanism of ochratoxin A carcinogenicity. Chem. Res. Toxicol. 2012, 25, 252–262. [Google Scholar]
- European Commission. Commission regulation (EC) No. 123/2005 of 26 January 2005 amending regulation (EC) No. 466/2001 as regards ochratoxin A. Off. J. Eur. Union. 2005, L25, 3–5.
- Duarte, S.; Bento, J.; Pena, A.; Lino, C.; Delerue-Matos, C.; Oliva-Teles, T.; Morais, S.; Correia, M.; Oliveira, M.; Alves, M. Monitoring of ochratoxin A exposure of the Portuguese population through a nationwide urine survey—Winter 2007. Sci. Total Environ. 2010, 408, 1195–1198. [Google Scholar]
- Rubert, J.; Sebastià, N.; Soriano, J.; Soler, C.; Mañes, J. One-year monitoring of aflatoxins and ochratoxin A in tiger-nuts and their beverages. Food Chem. 2011, 127, 822–826. [Google Scholar]
- Esti, M.; Benucci, I.; Liburdi, K.; Acciaro, G. Monitoring of ochratoxin A fate during alcoholic fermentation of wine-must. Food Control 2012, 27, 53–56. [Google Scholar]
- Sauceda-Friebe, J.C.; Karsunke, X.Y.; Vazac, S.; Biselli, S.; Niessner, R.; Knopp, D. Regenerable immuno-biochip for screening ochratoxin A in green coffee extract using an automated microarray chip reader with chemiluminescence detection. Anal. Chim. Acta 2011, 689, 234–242. [Google Scholar]
- Chen, J.; Fang, Z.; Liu, J.; Zeng, L. A simple and rapid biosensor for ochratoxin A based on a structure-switching signaling aptamer. Food Control 2012, 25, 555–560. [Google Scholar]
- Zamfir, L.-G.; Geana, I.; Bourigua, S.; Rotariu, L.; Bala, C.; Errachid, A.; Jaffrezic-Renault, N. Highly sensitive label-free immunosensor for ochratoxin A based on functionalized magnetic nanoparticles and EIS/SPR detection. Sens. Actuators B 2011, 159, 178–184. [Google Scholar] [CrossRef]
- Yu, F.-Y.; Vdovenko, M.M.; Wang, J.-J.; Sakharov, I.Y. Comparison of enzyme-linked immunosorbent assays with chemiluminescent and colorimetric detection for the determination of ochratoxin A in food. J. Agric. Food Chem. 2011, 59, 809–813. [Google Scholar] [CrossRef]
- Barthelmebs, L.; Jonca, J.; Hayat, A.; Prieto-Simon, B.; Marty, J.-L. Enzyme-linked aptamer assays (ELAAs), based on a competition format for a rapid and sensitive detection of ochratoxin A in wine. Food Control 2011, 22, 737–743. [Google Scholar] [CrossRef]
- Heurich, M.; Kadir, M.K.A.; Tothill, I.E. An electrochemical sensor based on carboxymethylated dextran modified gold surface for ochratoxin A analysis. Sens. Actuators B 2011, 156, 162–168. [Google Scholar] [CrossRef] [Green Version]
- Bonel, L.; Vidal, J.C.; Duato, P.; Castillo, J.R. An electrochemical competitive biosensor for ochratoxin A based on a DNA biotinylated aptamer. Biosens. Bioelectron. 2011, 26, 3254–3259. [Google Scholar] [CrossRef]
- Tothill, I.E. Biosensors and nanomaterials and their application for mycotoxin determination. World Mycotoxin J. 2011, 4, 361–374. [Google Scholar] [CrossRef] [Green Version]
- Mirsky, V.M.; Yatsimirsky, A. Artificial Receptors for Chemical Sensors; Wiley-VCH: Berlin, Germany, 2010. [Google Scholar]
- Kuchelmeister, H.Y.; Schmuck, C. Molecular Recognition of Oligopeptides and Protein Surfaces. In Designing Receptors for the Next Generation of Biosensors; Springer: Berlin, Germany, 2013; pp. 67–84. [Google Scholar]
- Van Regenmortel, M. Synthetic peptides versus natural antigens in immunoassays. Ann. Biol. Clin. 1993, 51, 39–41. [Google Scholar]
- Tothill, I.E. Peptides as Molecular Receptors. In Recognition Receptors in Biosensors; Zourob, M., Ed.; Springer: Berlin, Germany, 2010; pp. 249–274. [Google Scholar]
- Giraudi, G.; Anfossi, L.; Baggiani, C.; Giovannoli, C.; Tozzi, C. Solid-phase extraction of ochratoxin A from wine based on a binding hexapeptide prepared by combinatorial synthesis. J. Chromatogr. A 2007, 1175, 174–180. [Google Scholar] [CrossRef]
- Tozzi, C.; Anfossi, L.; Giraudi, G.; Giovannoli, C.; Baggiani, C.; Vanni, A. Chromatographic characterisation of an estrogen-binding affinity column containing tetrapeptides selected by a combinatorial-binding approach. J. Chromatogr. A 2002, 966, 71–79. [Google Scholar] [CrossRef]
- Tozzi, C.; Anfossi, L.; Baggiani, C.; Giovannoli, C.; Giraudi, G. A combinatorial approach to obtain affinity media with binding properties towards the aflatoxins. Anal. Bioanal. Chem. 2003, 375, 994–999. [Google Scholar]
- Clark, M.; Cramer, R.D.; Van Opdenbosch, N. Validation of the general purpose Tripos 5.2 force field. J. Comput. Chem. 2004, 10, 982–1012. [Google Scholar]
- Richon, A.B. An introduction to molecular modeling. Mathematech 1994, 1, 83. [Google Scholar]
- Böhm, H.-J. The computer program LUDI: A new method for the de novo design of enzyme inhibitors. J. Comput. Aided Mol. Des. 1992, 6, 61–78. [Google Scholar] [CrossRef]
- Chianella, I.; Lotierzo, M.; Piletsky, S.A.; Tothill, I.E.; Chen, B.; Karim, K.; Turner, A.P. Rational design of a polymer specific for microcystin-LR using a computational approach. Anal. Chem. 2002, 74, 1288–1293. [Google Scholar] [CrossRef]
- Subrahmanyam, S.; Piletsky, S.A.; Piletska, E.V.; Chen, B.; Karim, K.; Turner, A.P. “Bite-and-Switch” approach using computationally designed molecularly imprinted polymers for sensing of creatinine. Biosens. Bioelectron. 2001, 16, 631–637. [Google Scholar] [CrossRef]
- Chothia, C.; Janin, J. Principles of protein-protein recognition. Nature 1975, 256, 705. [Google Scholar] [CrossRef]
- Payne, A.; Glen, R.C. Molecular recognition using a binary genetic search algorithm. J. Mol. Graph. 1993, 11, 74–91. [Google Scholar] [CrossRef]
- Keskin, O.; Gursoy, A.; Ma, B.; Nussinov, R. Principles of protein-protein interactions: What are the preferred ways for proteins to interact? Chem. Rev. 2008, 108, 1225. [Google Scholar] [CrossRef]
- Chu, F.S. Interaction of ochratoxin A with bovine serum albumin. Arch. Biochem. Biophys. 1971, 147, 359–366. [Google Scholar] [CrossRef]
- Il’ichev, Y.V.; Perry, J.L.; Simon, J.D. Interaction of ochratoxin A with human serum albumin. Preferential binding of the dianion and pH effects. J. Phys. Chem. B 2002, 106, 452–459. [Google Scholar] [CrossRef]
- Karlsson, R.; Fält, A. Experimental design for kinetic analysis of protein-protein interactions with surface plasmon resonance biosensors. J. Immunol. Methods 1997, 200, 121–133. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Heurich, M.; Altintas, Z.; Tothill, I.E. Computational Design of Peptide Ligands for Ochratoxin A. Toxins 2013, 5, 1202-1218. https://doi.org/10.3390/toxins5061202
Heurich M, Altintas Z, Tothill IE. Computational Design of Peptide Ligands for Ochratoxin A. Toxins. 2013; 5(6):1202-1218. https://doi.org/10.3390/toxins5061202
Chicago/Turabian StyleHeurich, Meike, Zeynep Altintas, and Ibtisam E. Tothill. 2013. "Computational Design of Peptide Ligands for Ochratoxin A" Toxins 5, no. 6: 1202-1218. https://doi.org/10.3390/toxins5061202
APA StyleHeurich, M., Altintas, Z., & Tothill, I. E. (2013). Computational Design of Peptide Ligands for Ochratoxin A. Toxins, 5(6), 1202-1218. https://doi.org/10.3390/toxins5061202