1. Introduction
The term “mycotoxin” incorporates all the various secondary metabolites of molds. Although the negative effects of mycotoxins have been described since the middle ages (15–16th centuries [
1]) scientists only began to engage in the identification and monitoring of specific hazardous compounds in the from the mid-20th century. To date, more than 500 mycotoxins are known, but only a few dozen of these may be found in food and animal feed in potentially dangerous quantities [
2]. These toxins are produced mainly by five genera of fungi:
Aspergillus,
Penicillium,
Fusarium,
Alternaria and
Claviceps [
3]. Most mycotoxins are very stable with respect to temperature and chemical exposure. Plant products are contaminated by mycotoxins directly as growing crops, whereas animal products contain mycotoxins assimilated from contaminated feeds. The complex toxic effects of mycotoxins pose a significant risk to human and animal health and necessitate their effective monitoring and control [
4,
5].
Currently, most countries around the world have established regulatory requirements for the maximal permissible residue levels (MPRLs) of mycotoxins in various foods and feed [
4,
6]. Because there are considerable variations in the degree of contamination and the probability of mycotoxins being present in concentrations not exceeding MPRLs, accurate and sensitive quantitative detection methods are of primary importance. The development of such methods is mainly focused on chromatography [
7,
8], chromatography coupled with mass spectrometry [
9,
10,
11,
12,
13] or immunoassays [
14,
15,
16,
17]. Chromatography and mass spectrometry require the use of complex and expensive equipment and are therefore mainly applied for confirmatory analysis. Immunoassay methods, among which the enzyme-linked immunosorbent assay (ELISA) is the most commonly used format [
18,
19], are much simpler in design, implemented using relatively inexpensive equipment, provide high productivity by allowing the simultaneous testing of tens of samples, and enable the detection of mycotoxins with high sensitivity and accuracy [
20].
However, common ELISA kits require that all immunochemical interactions take place at equilibrium (or close to equilibrium), thus providing good reproducibility. As a result, total assay duration for different kits varies from one hour to several hours. For example, the application of Aflatoxins B1 [AFB1] ELISA Test Kit (Krishgen Biosystems, Los Angeles, CA, USA) requires 60 min, Zearalenone (ZEN) ELISA Kit (Cusabio Biotech Co., Ltd., Wuhan, China)—70 min, IDetect Ochratoxin A ELISA Test Kit (Idlabs Biotechnology Inc., London, ON, Canada)—75 min, Total Aflatoxin ELISA Kit (EuroClone SpA, Milan, Italy)—80 min, Aflatoxins B1 in food kit (Diagnostic Automation, Inc., Calabasas, CA, USA)—150 min.
This method also requires the transition of ELISA to the kinetic regime, and this is associated with a decrease in the number of immune complexes generated and detected, and accordingly, produces less accurate assay results. For certain applications though, assay durations of several hours are often unacceptable. Many tasks involved in raw material control, process monitoring and final product testing require a more rapid testing turnaround time. However, known methods of rapid immunoassay such as immunochromatography are focused primarily on qualitative “yes–no” testing and cannot serve as an adequate substitute for ELISA.
The kinetics of the interactions in ELISA have been studied in a number of works, including both theoretical research, starting with the classic papers of Rodbard [
21,
22], and experimental research [
23,
24,
25,
26,
27]. However, the application of this knowledge towards the development of express immunotechniques is very limited. The shortening of the analysis duration to 10–50 min [
28,
29] has been described only for specific individual antigens based on empirical evaluation of the binding kinetics. In addition, a shift to the kinetic regime reduces (to a greater or lesser degree) the binding of detectable markers, which must be accompanied by additional solutions to retain acceptable accuracy of analysis results. Methodological solutions for express immunoassays are provided in commercial tests (see
www.neogen.com/foodsafety/fs_da_index.html as an example), but the underlying methods are not disclosed by manufacturers. Therefore, the question of the simultaneous control of several compounds in the kinetic regime, which is especially important for the monitoring of mycotoxin contamination, remains open. It remains unclear to what extent the kinetics of immunochemical interactions vary for different antigens, and whether their combination in a single assay protocol is possible.
Given the above, our aim was to study the possibility of rapid control of multiple mycotoxins using a unified protocol based on modified microplate ELISA. The unification of stages duration allows to carry out all floods/incubations/washings simultaneously in different wells of microplate containing immunoreactants of different specificity and by this way to obtain information about all controlled mycotoxins after one assay cycle. Three mycotoxins, aflatoxin B1 (AFB1), ochratoxin A (OTA), and zearalenone (ZEA), were selected as test compounds owing to their wide presence in food stuffs and the significant threat they pose to consumers [
10,
14,
15]. Efficiency of the developed assay was validated using corn samples as one from priority foodstuffs contaminated by mycotoxins [
30] and poultry processing samples due to importance of contamination control along food chain and a lack of corresponding investigations for different matrixes of animal origin [
31,
32].
2. Results and Discussion
2.1. Choice of Assay Format and Overall Optimization
Two approaches to analyte labeling are generally used in ELISA, namely direct and indirect labeling [
33]. The first proposes the use of direct complexes between the enzyme and specific antibodies or competing antigen, thus reducing the number of incubation steps in the course of the assay. However, this approach can be influenced by negative effects of the test sample matrix on the enzyme label and the risk of enzyme inactivation. In the second approach, the enzyme is introduced to the assay only once the immune complexes have been formed. This approach eliminates contact between the sample matrix and the label and allows the use of the same labeled reagent for multiple analytes. In the case of mycotoxins, organic solvents and a wide range of extracted substances may inactivate the enzyme label. We therefore chose the indirect labeling of antibodies for our assay.
The proposed kinetic ELISA was accomplished with the use of several additional reagents or steps, thereby enabling the effective incorporation of the label in the complexes to be detected: (1) the biotin-streptavidin interaction was exploited for the detection of primary antibodies, as this complex has a higher binding constant (10
15 M
−1) compared with the interaction between primary antibodies and anti-species antibodies [
34]; (2) specific primary antibodies were modified with a biotin ester containing a 14-atom spacer (biotinamidohexanoyl-6-aminohexanoic acid
N-hydroxysuccinimide ester) [
35,
36]; and (3) the streptavidin moiety was modified with a polymer that enabled the coupling of several peroxidase molecules [
37,
38].
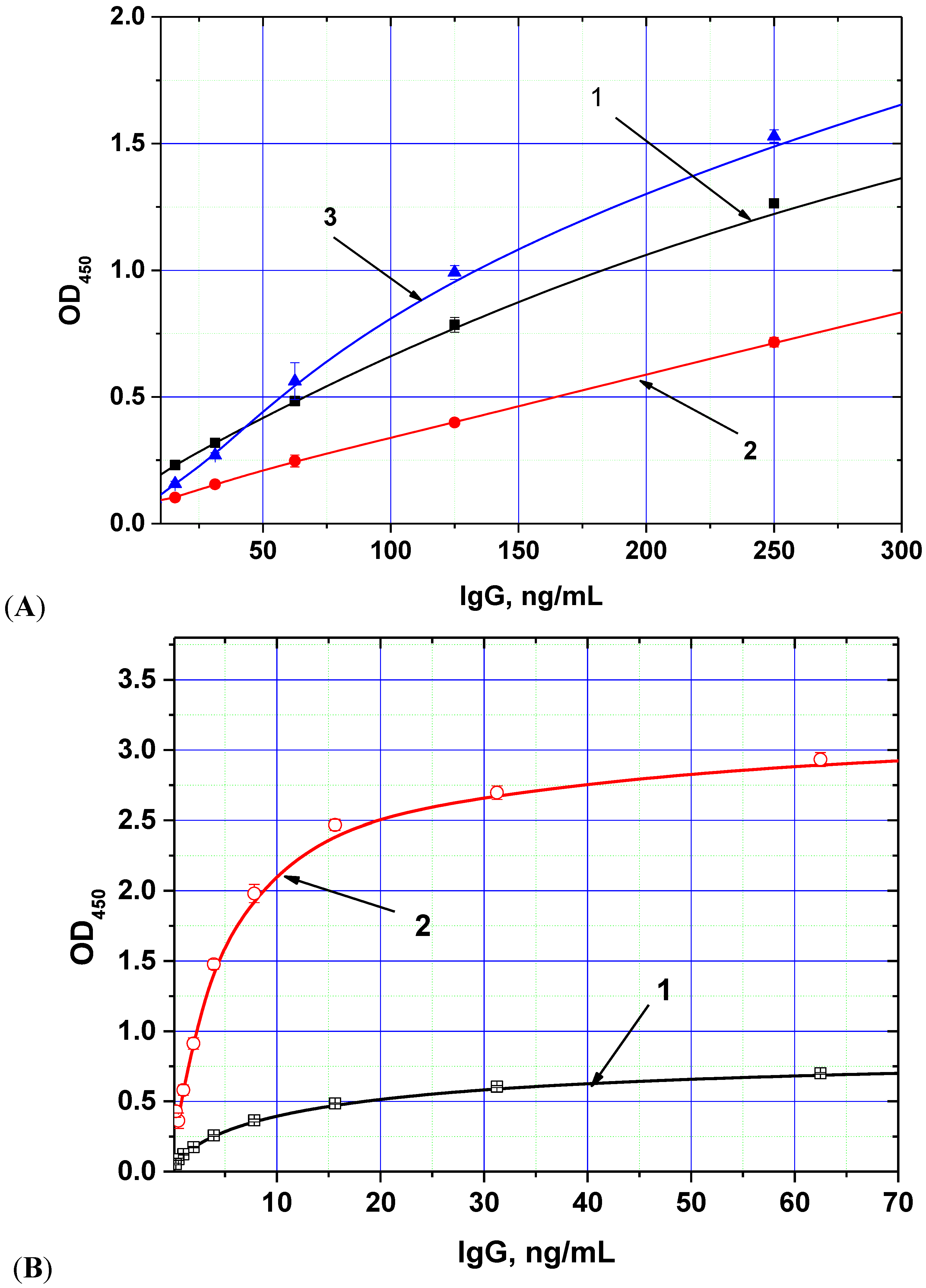
The chosen biotin derivative demonstrated two-fold increased binding to the labeled streptavidin compared with the usual biotin
N-hydroxysuccinimide ester (see
Figure 1A), whereas the streptavidin–polyperoxidase conjugate provided up to 5-fold increase of detected peroxidase activity (
Figure 1B).
Figure 1.
Dependences of optical density (OD) registered in the ELISA from the concentration of anti-AFB1 antibodies obtained for different methods of immune complex labeling. (
A) variants of peroxidase conjugate binding (1—antibody-biotin + antispecies antibody—peroxidase, 2—antibody-biotin + streptavidin-peroxidase, 3—antibody-additional bridge-biotin + streptavidin-peroxidase); (
B) variants of peroxidase conjugates (1—antispecies antibody—peroxidase, 2—streptavidin-polyperoxidase). AFB1-BSA conjugate was immobilized in microplate wells from 0.5 μg/mL; other parameters of the ELISA experiment are given in the
Experimental Section (data are presented for three replicates).
Figure 1.
Dependences of optical density (OD) registered in the ELISA from the concentration of anti-AFB1 antibodies obtained for different methods of immune complex labeling. (
A) variants of peroxidase conjugate binding (1—antibody-biotin + antispecies antibody—peroxidase, 2—antibody-biotin + streptavidin-peroxidase, 3—antibody-additional bridge-biotin + streptavidin-peroxidase); (
B) variants of peroxidase conjugates (1—antispecies antibody—peroxidase, 2—streptavidin-polyperoxidase). AFB1-BSA conjugate was immobilized in microplate wells from 0.5 μg/mL; other parameters of the ELISA experiment are given in the
Experimental Section (data are presented for three replicates).
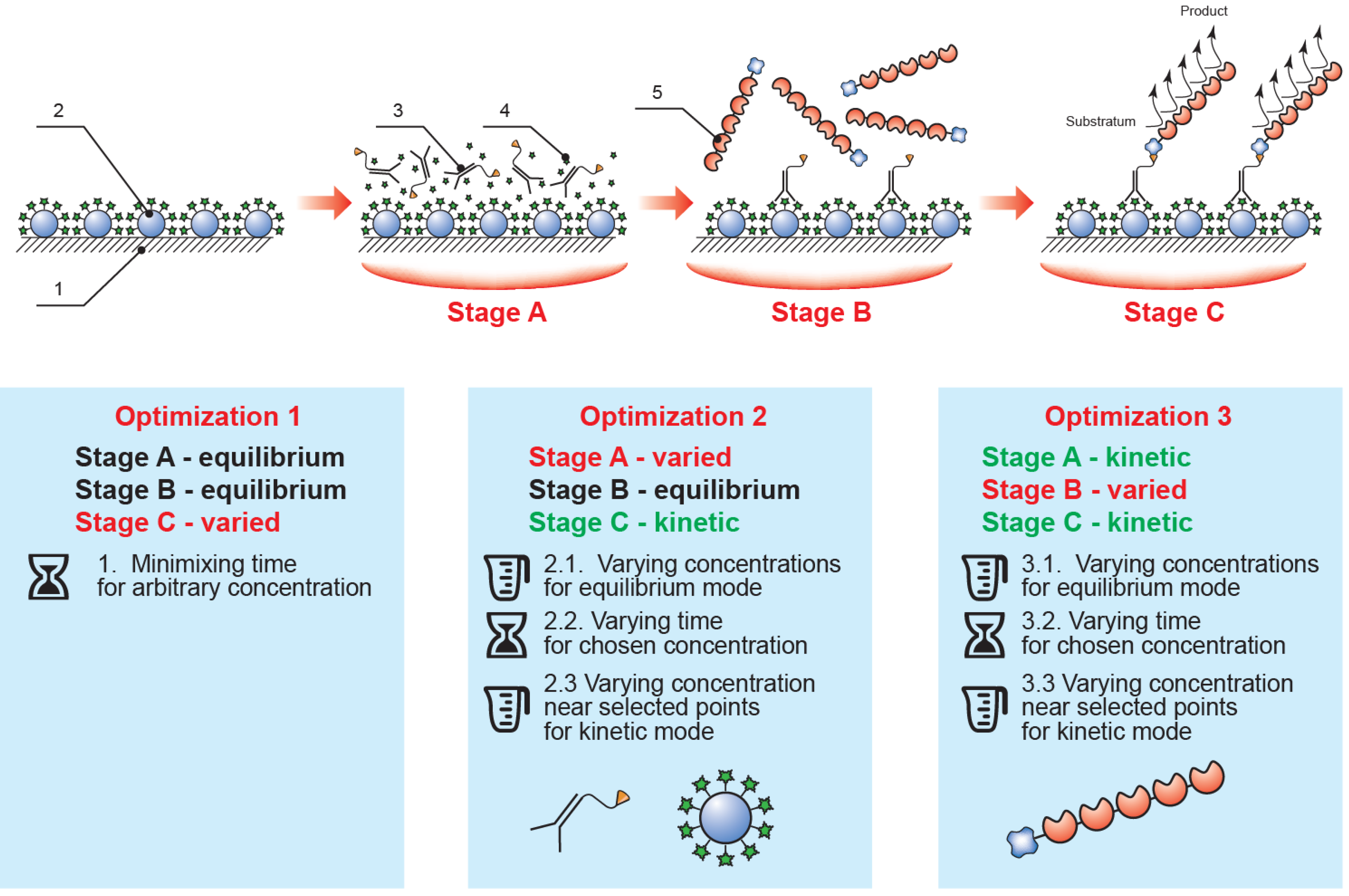
Thus, the implemented analysis included three sequential steps (see
Figure 2): (1) competitive interaction of the biotinylated antibody with immobilized mycotoxin–protein conjugate and free mycotoxin potentially contained in the sample; (2) interaction of the streptavidin–polyperoxidase conjugate with the biotin moiety of the formed immune complexes; and (3) detection of the catalytic activity of the bound enzyme label. Between each step the microplate was washed to remove any unbound components. The ELISA calibration curve was therefore determined by the conditions of the competitive interaction step (1), with the subsequent steps (2–3) merely allowing identification of complexes formed in the first step. This approach facilitates the manipulation of the parameters of the various stages to optimize them.
Figure 2.
The proposed rapid ELISA format and the optimization sequence employed (1, polystyrene microplate; 2, immobilized mycotoxin–protein conjugate; 3, specific antibody–biotin conjugate; 4, mycotoxin in the tested sample; 5, streptavidin–polyperoxidase conjugate; stage A, competitive immunochemical interaction; stage B, interaction of specific complexes with streptavidin-polyperoxidase; stage C, enzymatic reaction).
Figure 2.
The proposed rapid ELISA format and the optimization sequence employed (1, polystyrene microplate; 2, immobilized mycotoxin–protein conjugate; 3, specific antibody–biotin conjugate; 4, mycotoxin in the tested sample; 5, streptavidin–polyperoxidase conjugate; stage A, competitive immunochemical interaction; stage B, interaction of specific complexes with streptavidin-polyperoxidase; stage C, enzymatic reaction).
Development and optimization of the assay protocol was initiated from equilibrium conditions: steps (1) and (2), duration of 60 min; step (3) duration of 20 min. We confirmed experimentally that any further increase in step duration did not lead to a significant increase in the signal amplitude of the ELISA, ODmax (which refers to the optical density at 450 nm recorded after carrying out the ELISA in the absence of target analyte in the sample).
Next, the enzymatic step was optimized. We selected saturating concentrations of the immobilized mycotoxin–protein conjugate, specific biotinylated antibodies and the streptavidin–polyperoxidase conjugate. The final step duration was chosen to provide the achievement of OD
max in the range of 0.5–1.0, which permits quantitative determination of the analyte with maximum accuracy [
39].
Following optimization, the concentrations of the reactants and the step durations were consistently set for steps (1) and (2), whereas for step (3), the ODmax values were controlled so they would remain within the optimal range of 0.5–1.0 to reach the lowest possible limit of analyte detection. To provide ELISA-like accuracy, the resulting ODmax after all optimizations should not fall below 0.5.
2.2. Reducing the Duration of the Enzymatic Reaction
In accordance with the above principles, the development of the rapid ELISA method was then initiated for the assay of the three mycotoxins, AFB1, OTA, and ZEA, under equilibrium mode. Analytical characteristics of the corresponding protocols are shown in
Table 1.
Table 1.
Analytical characteristics of mycotoxin ELISAs under equilibrium mode.
Table 1.
Analytical characteristics of mycotoxin ELISAs under equilibrium mode.
| Mycotoxin | Analysis time, min | Limit of detection, ng/mL | Working range, ng/mL | Maximum deviation *, % | Average deviation *, % |
|---|
| AFB1 | 140 | 0.1 | 0.1–1.0 | 12.3 | 3.4 |
| OTA | 4.0 | 4.0–120.0 | 11.6 | 4.2 |
| ZEA | 0.3 | 0.3–50.0 | 14.2 | 4.7 |
The duration of the enzymatic reaction was optimized as described above. The resulting experimental data (
Table 2) demonstrated that an 8 min incubation period was sufficient for ELISA of all three mycotoxins.
Table 2.
Dependence of ELISA signal amplitude (ODmax) on the duration of the enzymatic reaction.
Table 2.
Dependence of ELISA signal amplitude (ODmax) on the duration of the enzymatic reaction.
| Mycotoxins | Time, min |
|---|
| 5 | 8 | 12 | 15 |
|---|
| AFB1 | 0.32 | 0.64 | 0.82 | 0.89 |
| ОТА | 0.27 | 0.58 | 0.76 | 0.93 |
| ZEA | 0.44 | 0.73 | 0.92 | 0.87 |
2.3. Choice of Reagent Concentrations for Competitive Immune Interaction
Considering the first step of the assay, we initially studied the most efficient immobilization procedure for the mycotoxin conjugates. The essential issue was the buffer choice for immobilization. For this purpose, the common recommendations for ELISA propose sodium-carbonate buffers of varying ionic strength with a pH of 9.0–9.5 [
39]. However, this is based on data obtained using antibodies, and is therefore not automatically valid for other proteins that may have different isoelectric points, hydrophobicity, molecular weights, and other characteristics. Initial comparisons of the competitive curves obtained by immobilization of the conjugates in phosphate (pH 7.4) and carbonate (pH 9.5) buffers showed negligible differences in terms of signal amplitude and reproducibility (data not shown). The use of the same media for initial immobilization and subsequent incubations was also considered preferable because this minimizes the risk of desorption and/or conformational changes of the adsorbed proteins. For this reason, in our study, microplate preparation was carried out using 50 mM phosphate buffer, pH 7.4, containing 100 mM NaCl (PBS).
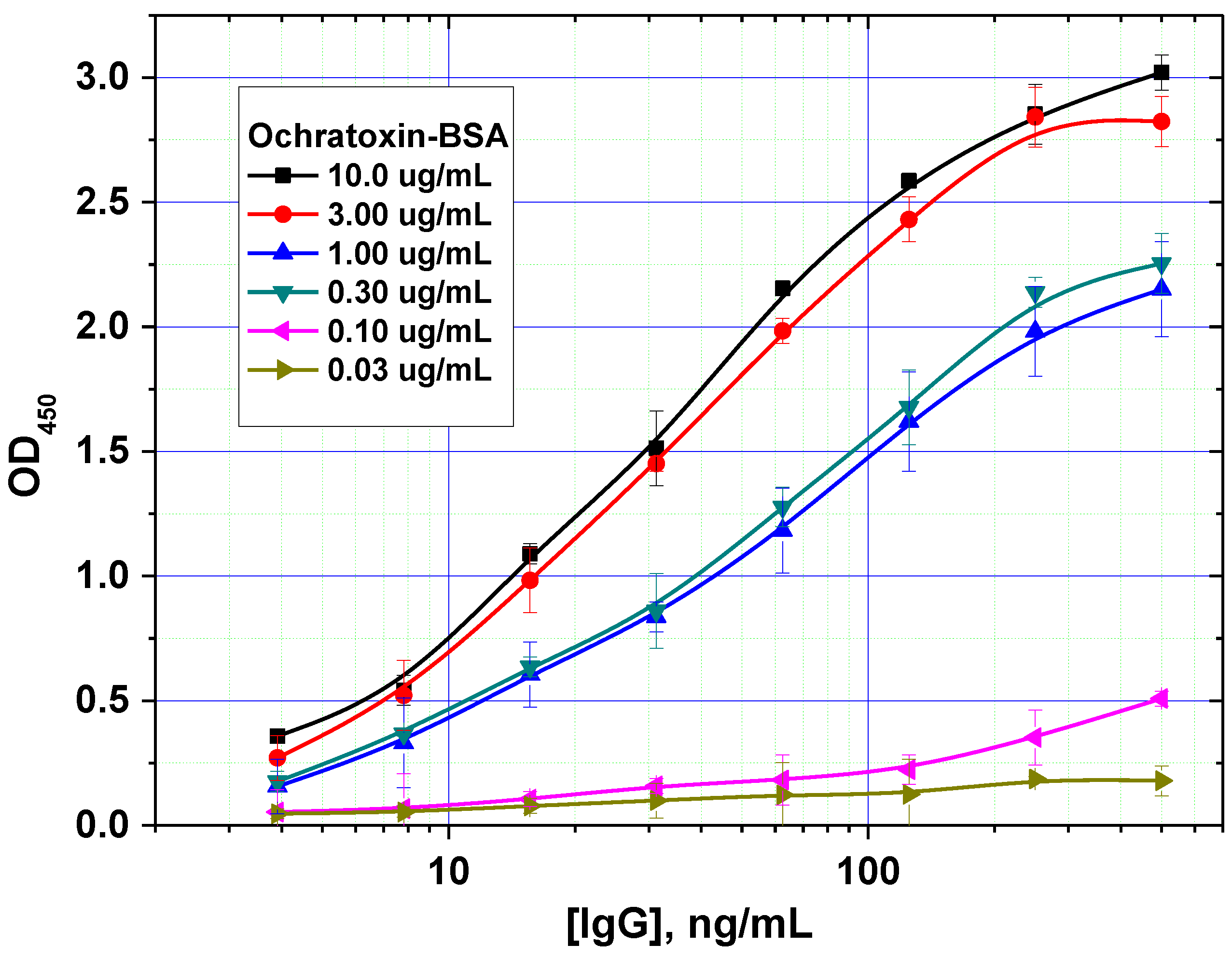
In addition, the optimal concentration of the immobilized mycotoxin–protein conjugate was determined. An example of such a comparison is shown in
Figure 3 for OTA. For this mycotoxin, a concentration of 0.3 µg/mL was shown to achieve the minimal limit of detection without loss of assay accuracy.
Figure 3.
Optimization of the concentration of immobilized ochratoxin A–bovine serum albumin (OTA–BSA) conjugate (data are presented for three replicates).
Figure 3.
Optimization of the concentration of immobilized ochratoxin A–bovine serum albumin (OTA–BSA) conjugate (data are presented for three replicates).
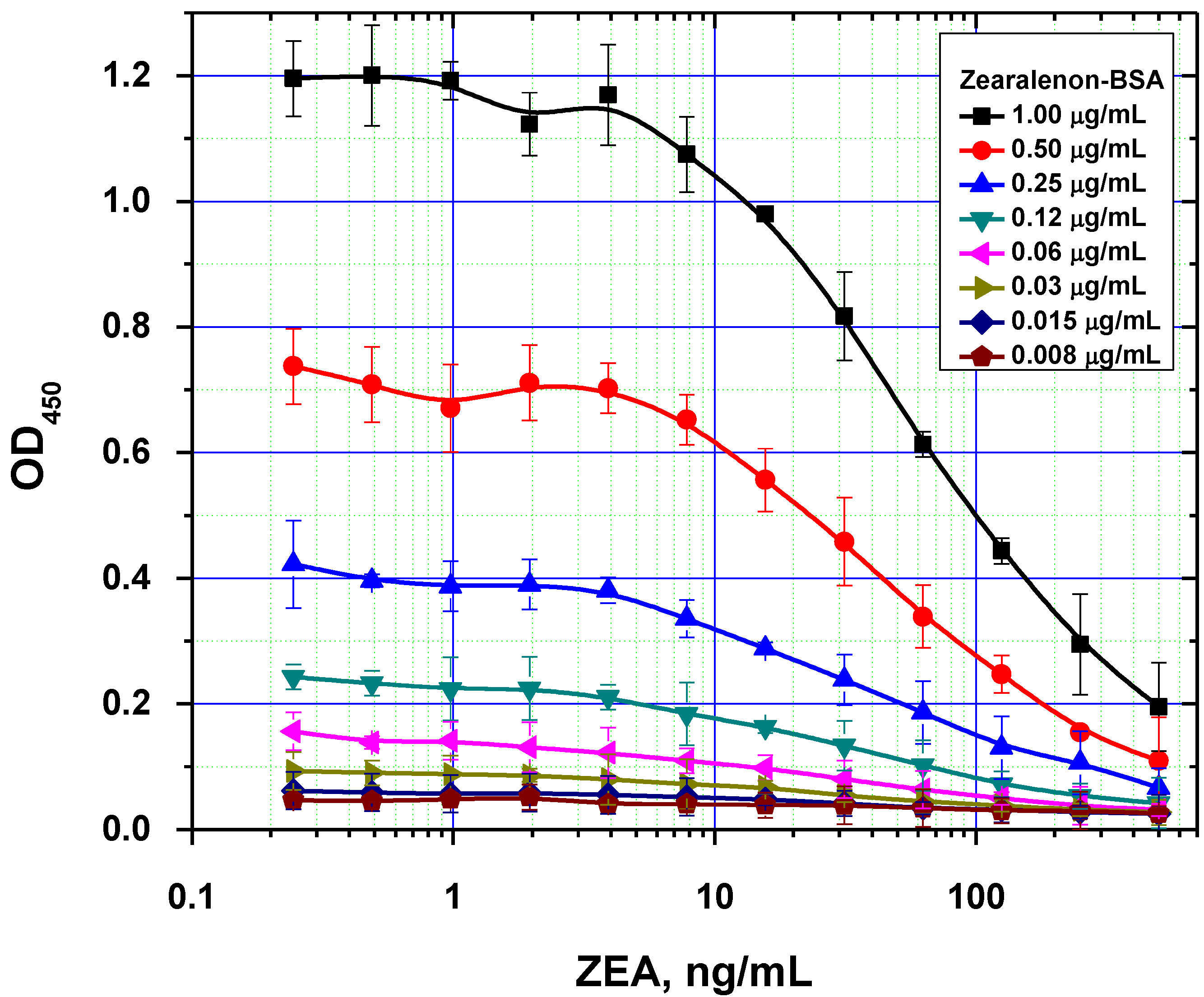
Figure 4.
Comparison of ELISA calibration curves for different concentrations of immobilized ZEA–BSA conjugate (data are presented for three replicates).
Figure 4.
Comparison of ELISA calibration curves for different concentrations of immobilized ZEA–BSA conjugate (data are presented for three replicates).
It was also demonstrated that excessive amounts of the immobilized reagent can result in two opposing effects. On one hand, it contributes to more efficient binding of the antibodies on the microplate surface. On the other hand, this negatively affects the analytical performance by increasing the detection limit, because of higher concentration of the competitors are necessary to inhibit antibody binding with solid phase (
Figure 4). As a result, the following values were chosen as optimal concentrations for rapid ELISAs under the equilibrium regime: 0.5, 0.3 and 1.0 µg/mL for AFB1-STI, OTA-BSA and ZEA-BSA, respectively.
2.4. Reducing the Duration of the Competitive Immunochemical Interaction
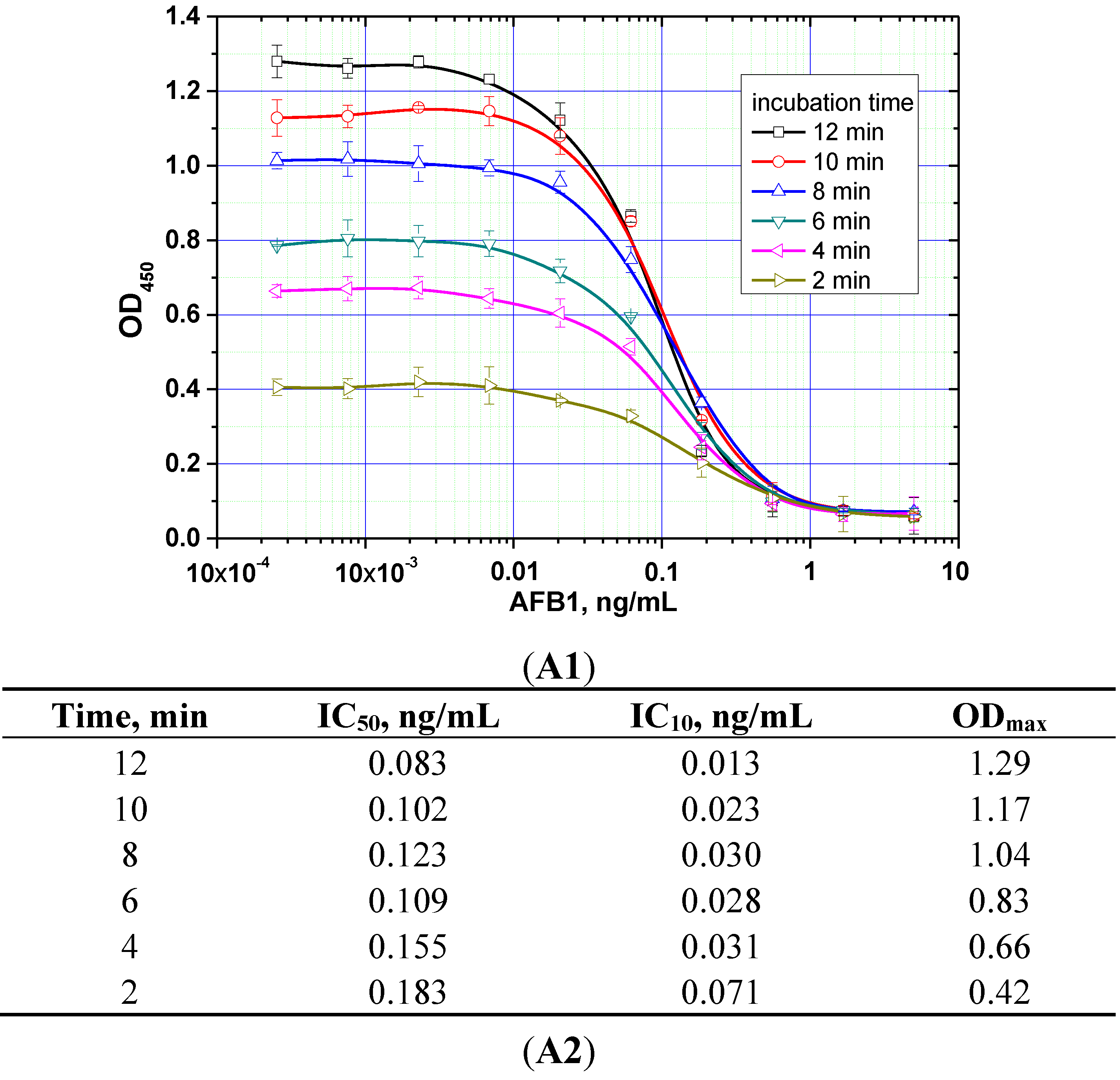
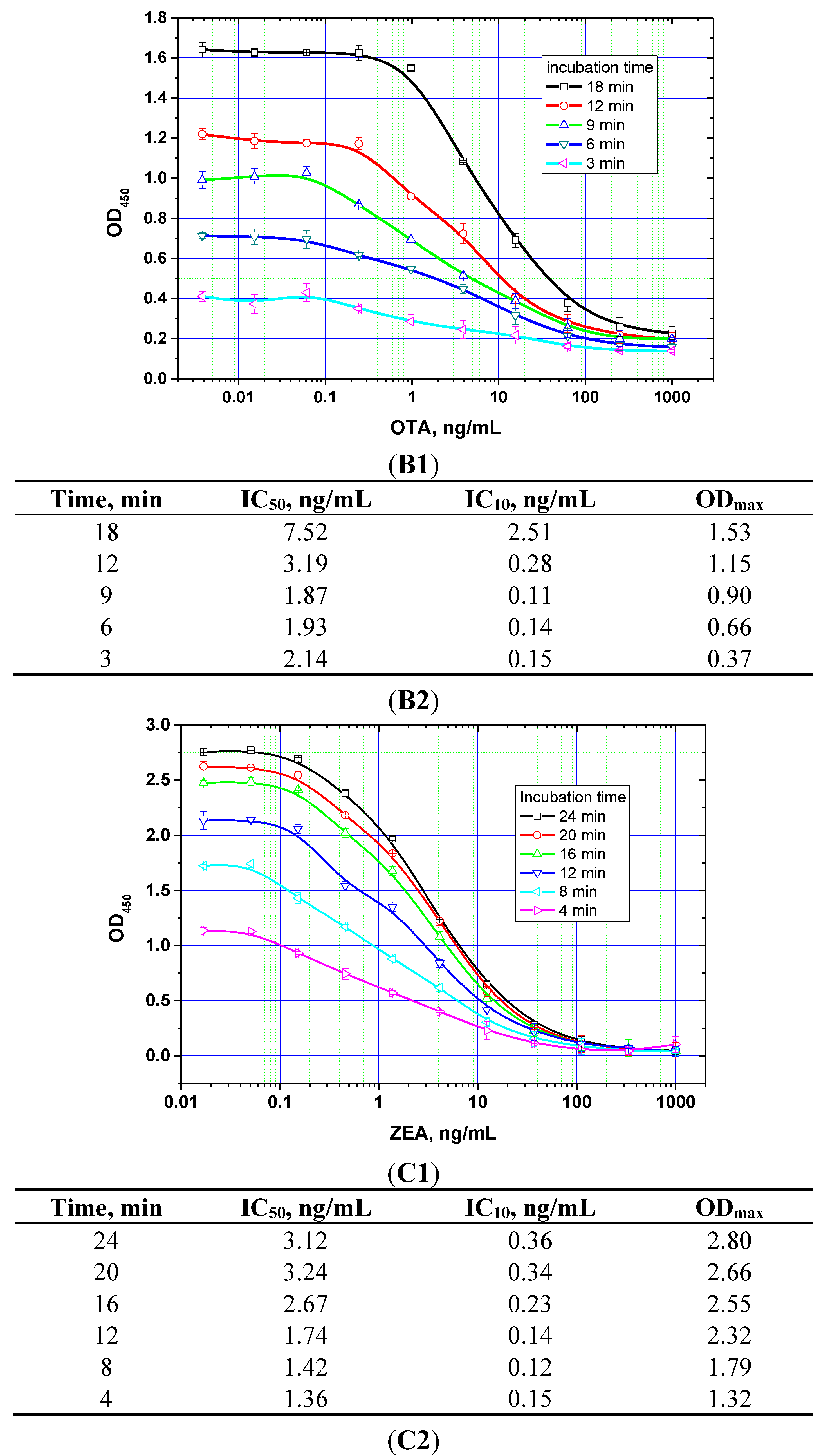
The duration of the competitive interaction was varied between 2 and 24 min (further increases in the duration did not result in reproducible increases in the signal amplitude). Examples of the corresponding experiments are shown in
Figure 5. For rapid ELISA of ZEA, increasing the duration of the competitive interaction resulted in a lower detection limit. With an incubation period up to 12 min, the detection limit reached <0.1 ng/mL (see
Figure 5C). However, even a 4-min incubation produced a signal amplitude >1.0. With a view towards optimized duration, the relevant optimal concentrations of the reagents were then selected for the kinetic ELISA method. Our optimization criterion was the choice of the minimum durations of the stages at which the signal amplitude is large enough (OD > 0.5) for the correct quantitative measurement of the assay results. Comparisons for the other two mycotoxins (see
Figure 5A,B) similarly informed the choice of optimal conditions for the competitive stage of ELISA, as summarized in
Table 3.
Figure 5.
Competitive curves of the immunoenzymatic determination of AFB1 (A1), OTA (B1), and ZEA (C1) and the analytical parameters there of (A2, B2, and C2, respectively) for different durations of the immunochemical interaction (IC50 for competitive ELISA is deemed the most accurate point for quantitative measurements, whereas IC10 is deemed the limit of detection). Data are presented for three replicates.
Figure 5.
Competitive curves of the immunoenzymatic determination of AFB1 (A1), OTA (B1), and ZEA (C1) and the analytical parameters there of (A2, B2, and C2, respectively) for different durations of the immunochemical interaction (IC50 for competitive ELISA is deemed the most accurate point for quantitative measurements, whereas IC10 is deemed the limit of detection). Data are presented for three replicates.
Table 3.
Selected conditions for the competitive immunochemical interaction and the resulting characteristics of the mycotoxin ELISAs.
Table 3.
Selected conditions for the competitive immunochemical interaction and the resulting characteristics of the mycotoxin ELISAs.
| Mycotoxins | Concentration of antibodies, ng/mL | Concentration of the mycotoxin–protein conjugate, µg/mL | Duration of the stage, min | Limit of detection, ng/mL | ODmax |
|---|
| AFB1 | 120 | 0.5 | 8 | 0.03 | 0.8 |
| OTA | 500 | 0.4 | 9 | 0.13 | 0.9 |
| ZEA | 100 | 1.0 | 4 | 0.15 | 1.3 |
2.5. Reducing the Duration of the Interaction between the Biotinylated Immune Complexes and the Streptavidin–Polyperoxidase Conjugate
The duration of the second assay step was varied between 3–18 min, while the first step had the relevant fixed duration as previously determined for each mycotoxin (see above). The parameters of the obtained competitive curves are summarized in
Table 4.
Table 4.
Signal amplitude and detection limit achieved using varying incubation periods for the interaction between the biotinylated immune complexes and streptavidin–polyperoxidase conjugate (data are presented for three replicates).
Table 4.
Signal amplitude and detection limit achieved using varying incubation periods for the interaction between the biotinylated immune complexes and streptavidin–polyperoxidase conjugate (data are presented for three replicates).
| Mycotoxins | Time, min |
|---|
| 3 | 6 | 9 | 12 | 18 |
|---|
| Signal amplitude, OD450/detection limit, ng/mL |
|---|
| AFB1 | 0.45/0.03 | 0.60/0.02 | 0.75/0.02 | 0.78/0.03 | 0.79/0.03 |
| OTA | 0.78/0.14 | 0.95/0.13 | 0.94/0.13 | 0.96/0.14 | 0.95/0.12 |
| ZEA | 0.80/0.21 | 0.88/0.11 | 0.92/0.23 | 1.10/0.30 | 1.15/0.30 |
The selection criterion was ODmax > 0.5. Accordingly, the duration of this stage for the rapid ELISA was chosen to be 6, 3, and 3 min for AFB1, OTA, and ZEA, respectively. It was also noted that reductions in the incubation time for this step had no effect on the observed detection limits.
2.6. Choice of Protocol for Kinetic Multianalysis of Mycotoxins
Once the reagent concentrations used at each step were optimized under the above determined incubation conditions, the assay protocols for kinetic ELISAs for the three individual mycotoxins could be finalized. The obtained results suggested the possibility of carrying out all three assays simultaneously, with a total assay time not exceeding 25 min (see
Table 5), and without any significant deterioration in analytical parameters compared with ELISA carried out in equilibrium mode.
Table 5.
Duration of rapid ELISA stages for the three mycotoxins studied.
Table 5.
Duration of rapid ELISA stages for the three mycotoxins studied.
| Mycotoxin | Competition interaction, min | Interaction with HRP conjugate, min | Enzymatic reaction, min | Total assay time *, min |
|---|
| AFB1 | 8 | 6 | 8 | 24 |
| ОТА | 9 | 3 | 8 | 22 |
| ZEA | 4 | 3 | 8 | 17 |
Combining these optimized protocols for application towards the simultaneous control of all three mycotoxins resulted in the following assay regime: 8-min duration for the competitive interaction, 6-min incubation with the peroxidase conjugate, and 8-min duration for the enzymatic reaction. This resulted in a total duration of 25 min for the multianalytical assay (including 3 min for auxiliary operations).
Transition to this regime did not lead to a significant change in the immunoassay characteristics of each analyte compared with the individual assays. Thus, a five-fold reduction in the total analysis time was obtainable relative to the equilibrium mode (see
Table 1 and
Table 6), without any significant deleterious effects on the analytical characteristics, yet with greatly increased speed and throughput.
Table 6.
Analytical characteristics of mycotoxin ELISA in the simultaneous kinetic regime.
Table 6.
Analytical characteristics of mycotoxin ELISA in the simultaneous kinetic regime.
| Mycotoxin | Analysis time, min | Limit of detection, ng/mL | Maximum deviation * | Average deviation * |
|---|
| AFB1 | 25 | 0.02 | 14.4% | 6.2% |
| OTA | 0.1 | 14.5% | 4.7% |
| ZEA | 0.25 | 15.0% | 6.8% |
Taking into consideration the extraction steps, the detection limits were equal to 0.24, 1.2 and 3 ng/g and working ranges were 0.25–10, 2–400, 5–500 ng/g for AFB1, OTA, and ZEA respectively.
2.7. Application of the Developed Methods for Testing of Real Samples
Raw extracts from poultry processing preparations and corn were taken for the testing of the developed methods. Mycotoxin extraction from was performed using standard procedures and a five-fold excess of a 70:30 methanol-water mixture. It should be noted that the used water-organic extraction is carried out by the same protocol thus allowing subsequent analysis of all three mycotoxins in the same extract.
Firstly, the absence of AFB1, OTA and ZEA in these matrixes was demonstrated using LC/fluorescence, LC/MS or HPLC methods [
40], the data were provided by University of Parma, Italy (Prof. A. Dossena) for poultry processing preparations and “Test-Pushchino”, Ltd., Russia (Dr. M. Voznyak) for corn samples. Then the extracts were spiked by known quantities of the mycotoxins. Initial extracts, spiked extracts and pure mycotoxins solutions were compared by the developed ELISA technique.
The calculations of the AFB1 content for spiked extracts based on the ELISA calibration curve give very close values (second column of the
Table 7 and
Table 8 for the AFB1 experiments) to the known added concentrations of the AFB1 (the first column). Besides, initial (not containing mycotoxins) extracts did not affect the recorded OD in the ELISA. The found value of AFB1 (basing on calibration curve for pure AFB1 solutions) was 0 ng/g for all repetitions.
Similar trends were demonstrated for two other studied mycotoxins (see
Table 7 and
Table 8 for OTA and ZEA) and confirmed reliable measurements of mycotoxins in real samples by the proposed ELISA techniques. Recovery of the added mycotoxins varied from 94.0% to 111.2%.
The average deviations did not exceed 10%, whereas the average recovery of AFB1, OTA, and ZEA were 98%, 102%, and 101%, respectively.
According to European Commission regulations [
41], the maximum permissible levels of AFB1, OTA, and ZEA in foodstuffs are 2, 2, and 20 ng/g respectively. The proposed system therefore provides sufficient assay sensitivity for the purposes of practical application.
Table 7.
Determination of mycotoxins in extracts of animal product samples.
Table 7.
Determination of mycotoxins in extracts of animal product samples.
| Added, ng/g | Found *, ng/g | Average deviation of found values, ng/g | Recovery, % |
|---|
| AFB1 |
| 8.4 | 8.2 | 0.2 | 97.6 |
| 3.4 | 3.3 | 0.2 | 97.1 |
| 1.0 | 1.0 | 0.1 | 100.0 |
| 0.3 | 0.3 | 0.1 | 100.0 |
| 0 | 0 | 0 | - |
| OTA |
| 120 | 118.2 | 0.7 | 98.5 |
| 32 | 35.5 | 0.6 | 111.2 |
| 8 | 7.5 | 0.1 | 94.0 |
| 2 | 1.9 | 0.1 | 96.0 |
| 0 | 0 | 0 | - |
| ZEA |
| 200 | 198.2 | 13.6 | 99.1 |
| 70 | 72.2 | 7.7 | 103.1 |
| 24 | 25.4 | 2.2 | 105.8 |
| 8 | 7.7 | 0.5 | 96.2 |
| 0 | 0 | 0 | - |
Table 8.
Determination of mycotoxins in extracts of corn.
Table 8.
Determination of mycotoxins in extracts of corn.
| Added, ng/g | Found *, ng/g | Average deviation of found values, ng/g | Recovery, % |
|---|
| AFB1 |
| 10 | 10.0 | 0.1 | 100.0 |
| 5 | 4.9 | 0.1 | 98.0 |
| 1 | 1.0 | 0.1 | 100.0 |
| 0 | 0 | 0 | - |
| OTA |
| 50 | 50.2 | 0.2 | 100.4 |
| 25 | 24.5 | 0.3 | 98.0 |
| 10 | 9.7 | 0.1 | 97.0 |
| 0 | 0 | 0 | - |
| ZEA |
| 100 | 104.1 | 5.1 | 104.1 |
| 50 | 54.2 | 4.3 | 108.4 |
| 10 | 9.6 | 1.6 | 96.0 |
| 0 | 0 | 0 | - |



 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



