
Triggering of Erythrocyte Death by Triparanol

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
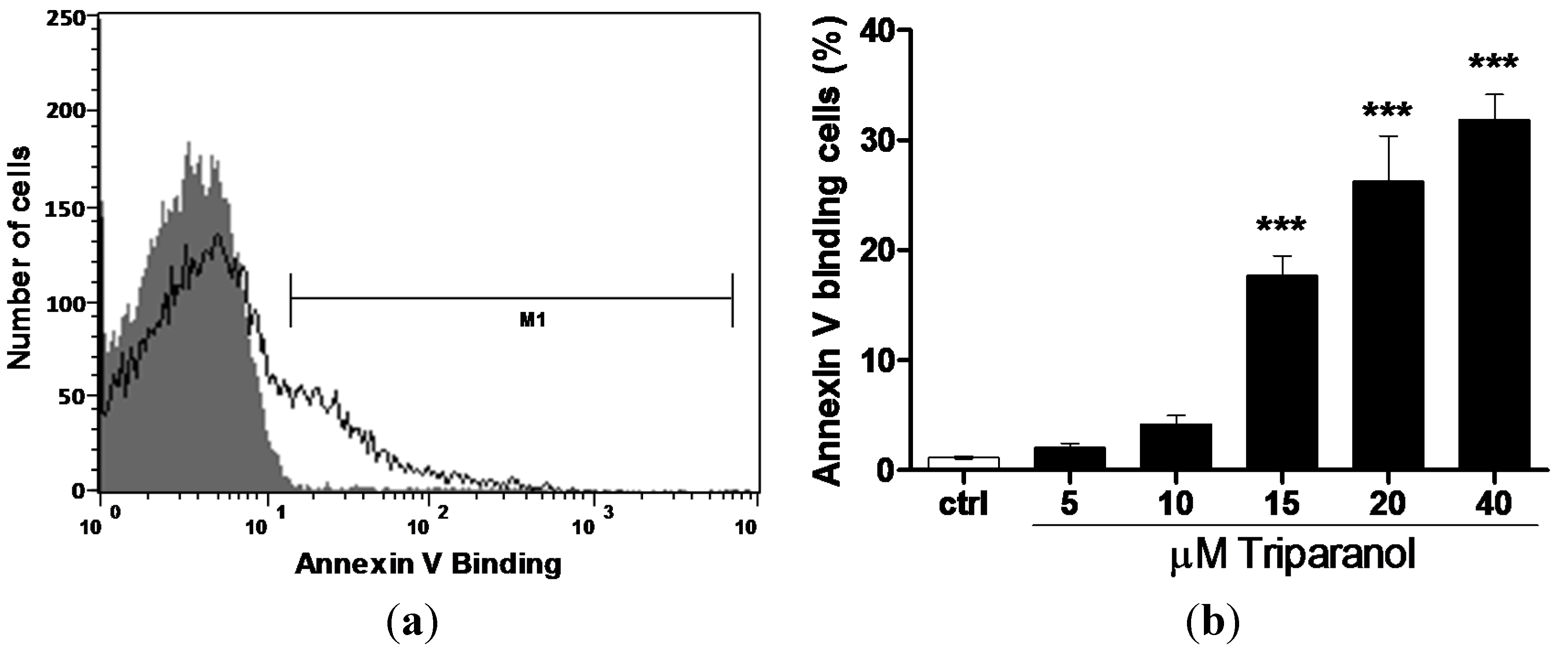
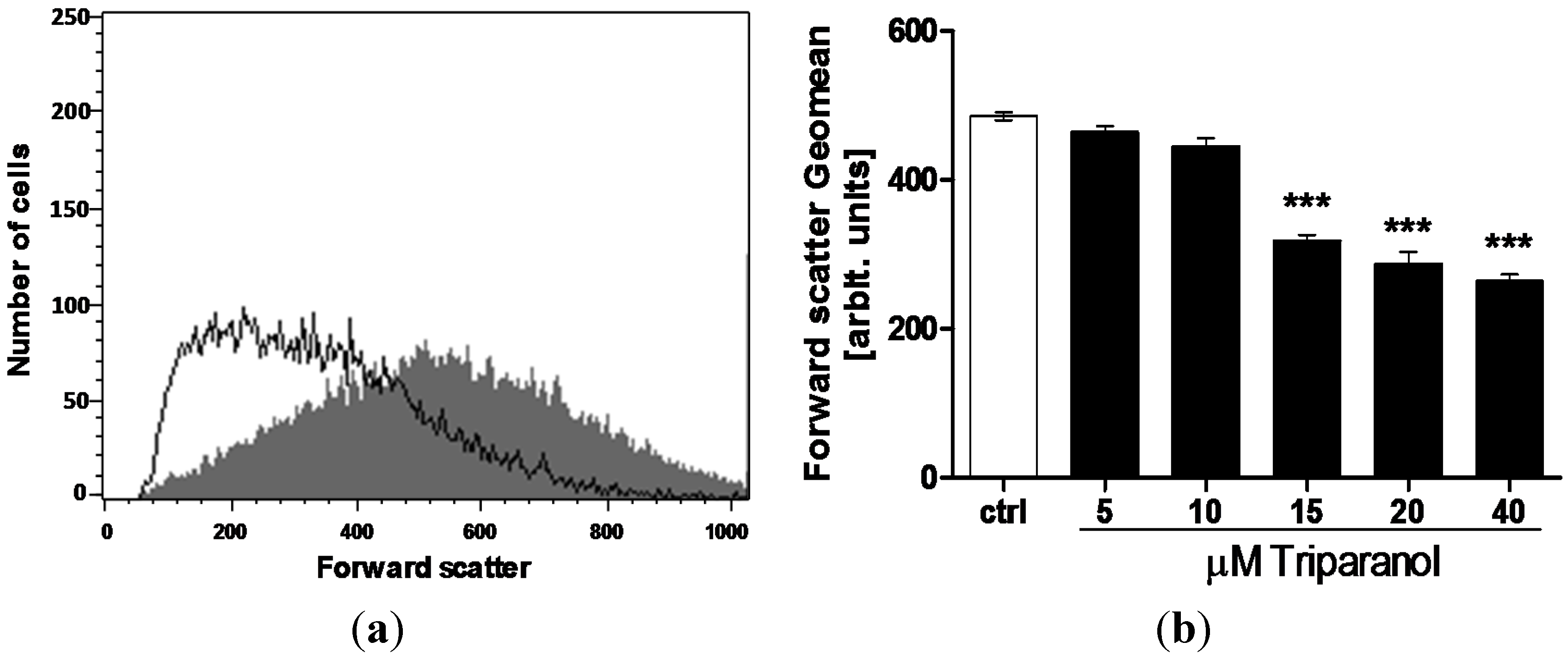
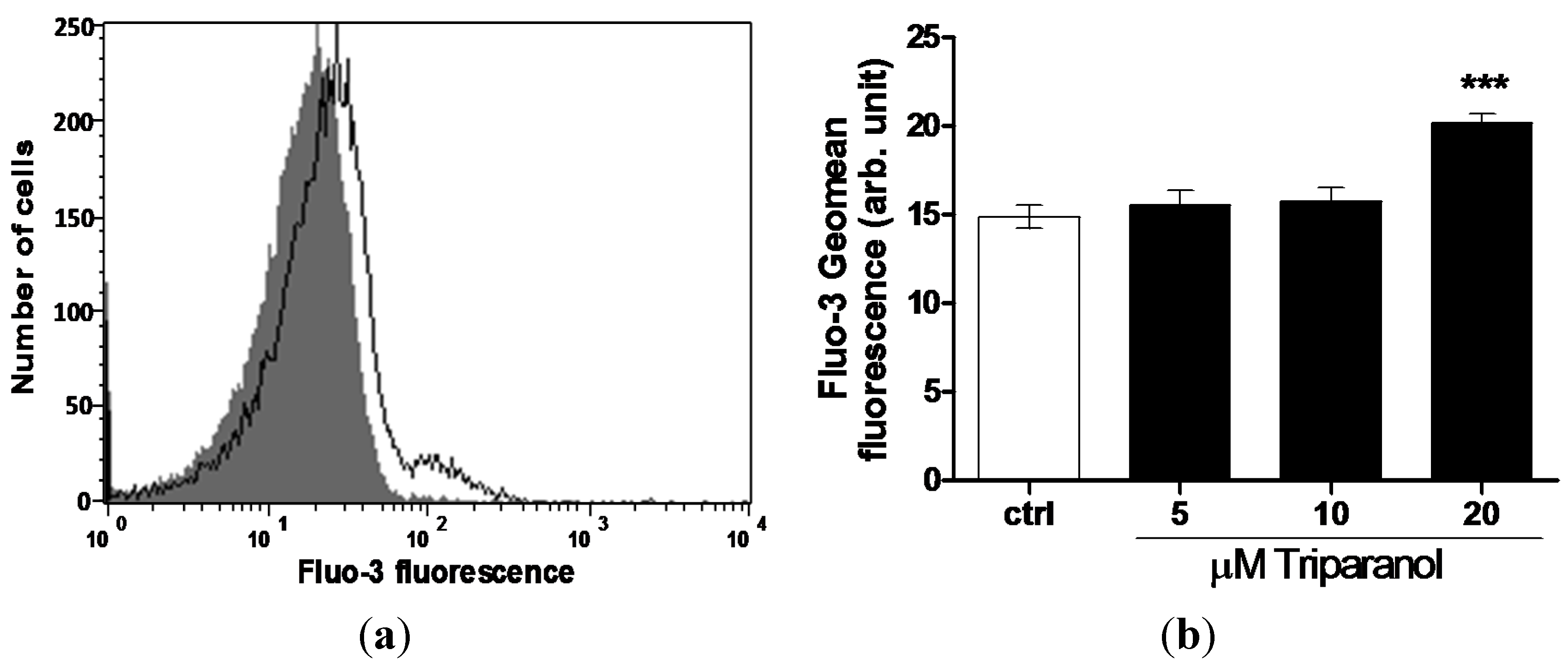
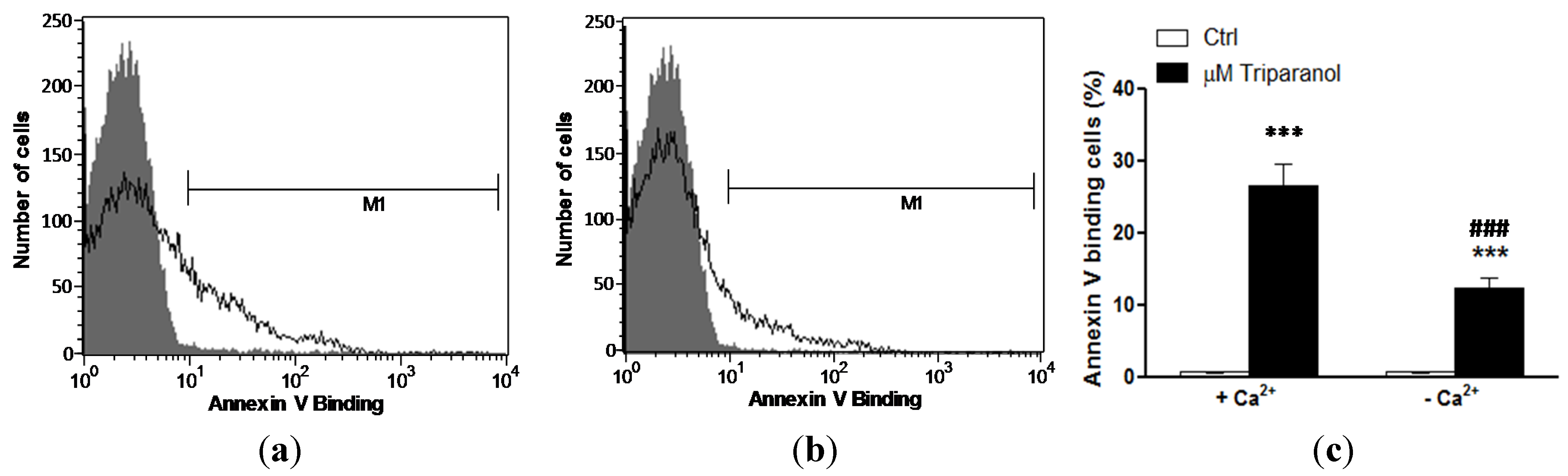
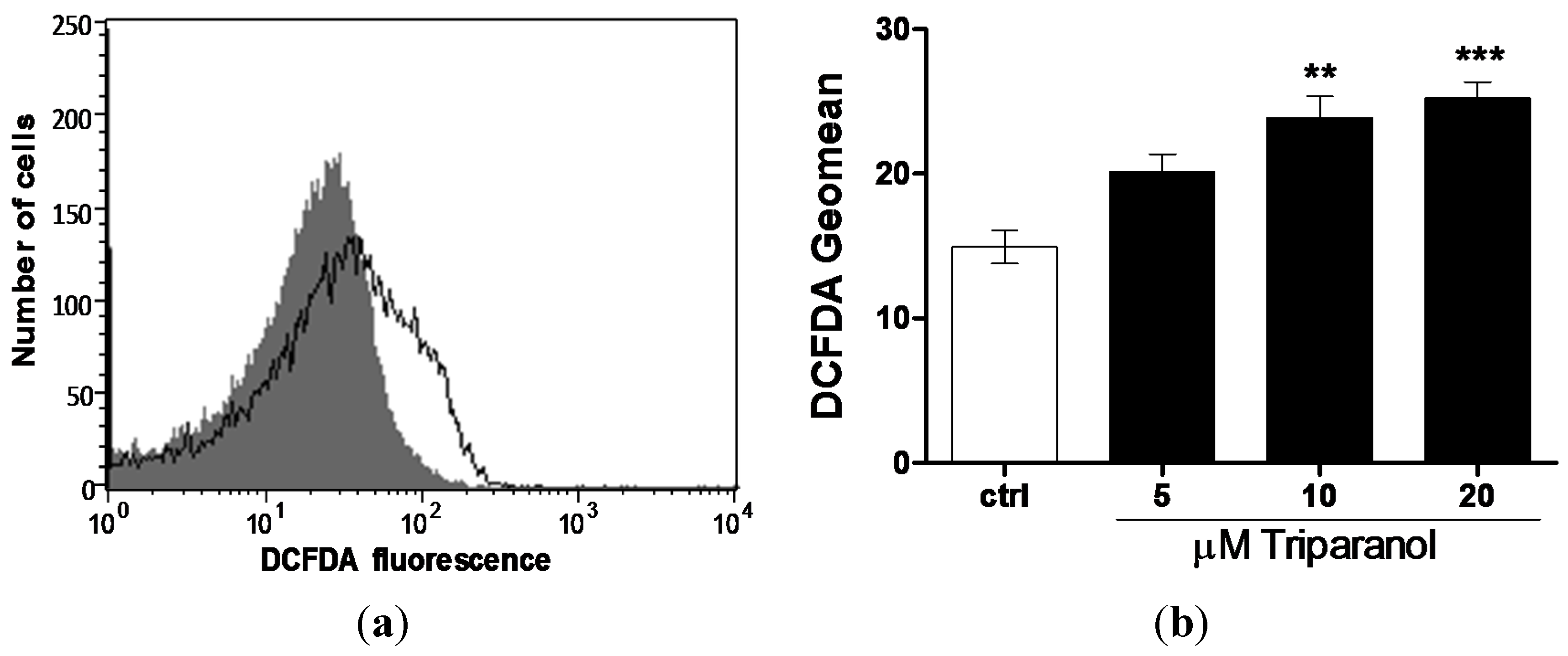
2. Results and Discussion





3. Experimental Section
3.1. Erythrocytes, Platelets, Solutions and Chemicals
3.2. Annexin-V-binding and Forward Scatter
3.3. Hemolysis
3.4. Intracellular Ca2+
3.5. Reactive Oxidant Species (ROS)
3.6. GSH Abundance
3.7. Statistics
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gofflot, F.; Hars, C.; Illien, F.; Chevy, F.; Wolf, C.; Picard, J.J.; Roux, C. Molecular mechanisms underlying limb anomalies associated with cholesterol deficiency during gestation: Implications of hedgehog signaling. Hum. Mol. Genet. 2003, 12, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Hihara, T.; Taniguchi, T.; Ueda, M.; Yoshinaga, T.; Miyamoto, N.; Sawada, K. Probucol and the cholesterol synthesis inhibitors simvastatin and triparanol regulate Iks channel function differently. Hum. Exp. Toxicol. 2013, 32, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Meyers, W.C.; Hanks, J.B.; Jakoi, L.; Quarfordt, S.; Jones, R.S. Selective biliary secretion of basal and glucagon-inhibited neutral sterol after triparanol administration. Surgery 1980, 88, 156–161. [Google Scholar] [PubMed]
- Roux, C.; Wolf, C.; Mulliez, N.; Gaoua, W.; Cormier, V.; Chevy, F.; Citadelle, D. Role of cholesterol in embryonic development. Am. J. Clin. Nutr. 2000, 71, 1270S–1279S. [Google Scholar] [PubMed]
- Bi, X.; Han, X.; Zhang, F.; He, M.; Zhang, Y.; Zhi, X.Y.; Zhao, H. Triparanol suppresses human tumor growth in vitro and in vivo. Biochem. Biophys. Res. Commun. 2012, 425, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Popjak, G.; Meenan, A.; Parish, E.J.; Nes, W.D. Inhibition of cholesterol synthesis and cell growth by 24(R,S),25-iminolanosterol and triparanol in cultured rat hepatoma cells. J. Biol. Chem. 1989, 264, 6230–6238. [Google Scholar] [PubMed]
- Tiet, T.D.; Hopyan, S.; Nadesan, P.; Gokgoz, N.; Poon, R.; Lin, A.C.; Yan, T.; Andrulis, I.L.; Alman, B.A.; Wunder, J.S. Constitutive hedgehog signaling in chondrosarcoma up-regulates tumor cell proliferation. Am. J. Pathol. 2006, 168, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.K.; Porter, J.A.; Young, K.E.; Beachy, P.A. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science 1998, 280, 1603–1607. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.P.; Kimura, T.; Yanagihara, T.; Sakoda, S. Calcium increase in mouse skeletal muscles by triparanol: A drug to induce myotonic dystrophy-like clinical manifestations. Neurosci. Lett. 1999, 272, 87–90. [Google Scholar] [CrossRef]
- Ghanbari-Azarnier, R.; Sato, S.; Wei, Q.; Al-Jazrawe, M.; Alman, B.A. Targeting stem cell behavior in desmoid tumors (aggressive fibromatosis) by inhibiting hedgehog signaling. Neoplasia 2013, 15, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Hopyan, S.; Nadesan, P.; Yu, C.; Wunder, J.; Alman, B.A. Dysregulation of hedgehog signalling predisposes to synovial chondromatosis. J. Pathol. 2005, 206, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Wei, Q.; Han, I.; Sato, S.; Ghanbari-Azarnier, R.; Whetstone, H.; Poon, R.; Hu, J.; Zheng, F.; Zhang, P.; et al. Hedgehog and notch signaling regulate self-renewal of undifferentiated pleomorphic sarcomas. Cancer Res. 2012, 72, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, G.R.; Chapman, C.J.; Chipault, J.R.; Pfeiffer, D.R. Lipid composition and (Na++ K+)-ATPase activity in rat lens during triparanol-induced cataract formation. Biochim. Biophys. Acta 1981, 644, 1–12. [Google Scholar] [CrossRef]
- Lang, P.A.; Kaiser, S.; Myssina, S.; Wieder, T.; Lang, F.; Huber, S.M. Role of Ca2+-activated K+ channels in human erythrocyte apoptosis. Am. J. Physiol. Cell. Physiol. 2003, 285, C1553–C1560. [Google Scholar] [CrossRef] [PubMed]
- Qadri, S.M.; Mahmud, H.; Lang, E.; Gu, S.; Bobbala, D.; Zelenak, C.; Jilani, K.; Siegfried, A.; Foller, M.; Lang, F. Enhanced suicidal erythrocyte death in mice carrying a loss-of-function mutation of the adenomatous polyposis coli gene. J. Cell. Mol. Med. 2012, 16, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Towhid, S.T.; Mia, S.; Pakladok, T.; Alesutan, I.; Borst, O.; Gawaz, M.; Gulbins, E.; Lang, F. Sphingomyelinase-induced adhesion of eryptotic erythrocytes to endothelial cells. Am. J. Physiol. Cell. Physiol. 2012, 303, C991–C999. [Google Scholar] [CrossRef] [PubMed]
- Lau, I.P.; Chen, H.; Wang, J.; Ong, H.C.; Leung, K.C.; Ho, H.P.; Kong, S.K. In vitro effect of CTAB- and PEG-coated gold nanorods on the induction of eryptosis/erythroptosis in human erythrocytes. Nanotoxicology 2012, 6, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Maellaro, E.; Leoncini, S.; Moretti, D.; Del Bello, B.; Tanganelli, I.; De Felice, C.; Ciccoli, L. Erythrocyte caspase-3 activation and oxidative imbalance in erythrocytes and in plasma of type 2 diabetic patients. Acta Diabetol. 2013, 50, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Jilani, K.; Lang, F. Carmustine-induced phosphatidylserine translocation in the erythrocyte membrane. Toxins 2013, 5, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Vota, D.M.; Maltaneri, R.E.; Wenker, S.D.; Nesse, A.B.; Vittori, D.C. Differential erythropoietin action upon cells induced to eryptosis by different agents. Cell. Biochem. Biophys. 2013, 65, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Zappulla, D. Environmental stress, erythrocyte dysfunctions, inflammation, and the metabolic syndrome: Adaptations to CO2 increases? J. Cardiometab. Syndr. 2008, 3, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Lupescu, A.; Jilani, K.; Zbidah, M.; Lang, F. Patulin-induced suicidal erythrocyte death. Cell. Physiol. Biochem. 2013, 32, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Zoubi, K.A.; Theurer, M.; Lang, F. Effect of dermaseptin on erythrocytes. Basic Clin. Pharmacol. Toxicol. 2013, 113, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.S.; Langer, H.; Abed, M.; Voelkl, J.; Lang, F. The uremic toxin acrolein promotes suicidal erythrocyte death. Kidney Blood Press Res. 2013, 37, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Ghashghaeinia, M.; Cluitmans, J.C.; Toulany, M.; Saki, M.; Koberle, M.; Lang, E.; Dreischer, P.; Biedermann, T.; Duszenko, M.; Lang, F.; et al. Age sensitivity of NFkB abundance and programmed cell death in erythrocytes induced by NFkB inhibitors. Cell. Physiol. Biochem. 2013, 32, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Alzoubi, K.; Honisch, S.; Abed, M.; Lang, F. Triggering of suicidal erythrocyte death by penta-o-galloyl-beta-d-glucose. Toxins 2014, 6, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Jilani, K.; Enkel, S.; Bissinger, R.; Almilaji, A.; Abed, M.; Lang, F. Fluoxetine induced suicidal erythrocyte death. Toxins 2013, 5, 1230–1243. [Google Scholar] [CrossRef] [PubMed]
- Lupescu, A.; Bissinger, R.; Jilani, K.; Lang, F. Triggering of suicidal erythrocyte death by celecoxib. Toxins 2013, 5, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Lang, E.; Modicano, P.; Bissinger, R.; Faggio, C.; Abed, M.; Lang, F. Effect of nitazoxanide on erythrocytes. Basic Clin. Pharmacol. Toxicol. 2014, 114, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Oswald, G.; Alzoubi, K.; Abed, M.; Lang, F. Stimulation of suicidal erythrocyte death by ribavirin. Basic Clin. Pharmacol. Toxicol. 2014, 114, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Lupescu, A.; Bissinger, R.; Herrmann, T.; Oswald, G.; Jilani, K.; Lang, F. Induction of suicidal erythrocyte death by novobiocin. Cell. Physiol. Biochem. 2014, 33, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Feger, M.; Alzoubi, K.; Pakladok, T.; Frauenfeld, L.; Geiger, C.; Towhid, S.T.; Lang, F. Sensitization of erythrocytes to suicidal erythrocyte death following water deprivation. Kidney Blood Press Res. 2013, 37, 567–578. [Google Scholar] [PubMed]
- Alzoubi, K.; Calabròa, S.; Bissinger, R.; Abed, M.; Faggio, C.; Lang, F. Stimulation of suicidal erythrocyte death by artesunate. Cell. Physiol. Biochem. 2014, 34, 2232–2244. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Bissinger, R.; Lang, F. Mitoxantrone-induced suicidal erythrocyte death. Cell. Physiol. Biochem. 2014, 34, 1756–1767. [Google Scholar] [CrossRef] [PubMed]
- Bissinger, R.; Fischer, S.; Jilani, K.; Lang, F. Stimulation of erythrocyte death by phloretin. Cell. Physiol. Biochem. 2014, 34, 2256–2265. [Google Scholar] [CrossRef] [PubMed]
- Bissinger, R.; Lupescu, A.; Zelenak, C.; Jilani, K.; Lang, F. Stimulation of eryptosis by cryptotanshinone. Cell. Physiol. Biochem. 2014, 34, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Bissinger, R.; Modicano, P.; Frauenfeld, L.; Lang, E.; Jacobi, J.; Faggio, C.; Lang, F. Estramustine-induced suicidal erythrocyte death. Cell. Physiol. Biochem. 2013, 32, 1426–1436. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, J.; Lang, E.; Bissinger, R.; Frauenfeld, L.; Modicano, P.; Faggio, C.; Abed, M.; Lang, F. Stimulation of erythrocyte cell membrane scrambling by mitotane. Cell. Physiol. Biochem. 2014, 33, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Lupescu, A.; Bissinger, R.; Warsi, J.; Jilani, K.; Lang, F. Stimulation of erythrocyte cell membrane scrambling by gedunin. Cell. Physiol. Biochem. 2014, 33, 1838–1848. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.; Bissinger, R.; Calabro, S.; Faggio, C.; Jilani, K.; Lang, F. Aristolochic acid induced suicidal erythrocyte death. Kidney Blood Press Res. 2014, 39, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Tesoriere, L.; Attanzio, A.; Allegra, M.; Cilla, A.; Gentile, C.; Livrea, M.A. Oxysterol mixture in hypercholesterolemia-relevant proportion causes oxidative stress-dependent eryptosis. Cell. Physiol. Biochem. 2014, 34, 1075–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voelkl, J.; Alzoubi, K.; Mamar, A.K.; Ahmed, M.S.; Abed, M.; Lang, F. Stimulation of suicidal erythrocyte death by increased extracellular phosphate concentrations. Kidney Blood Press Res. 2013, 38, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Xiang, Y.; Ran, Q.; Deng, X.; Xiao, Y.; Xiang, L.; Li, Z. Involvement of calcium, reactive oxygen species, and ATP in hexavalent chromium-induced damage in red blood cells. Cell. Physiol. Biochem. 2014, 34, 1780–1791. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D.; Avigan, J.; Feigelson, E.B. Effects of triparanol (MER-29) on cholesterol biosynthesis and on blood sterol levels in man. J. Clin. Invest. 1961, 40, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Artunc, F.; Alzoubi, K.; Honisch, S.; Baumann, D.; Foller, M.; Lang, F. Suicidal erythrocyte death in end-stage renal disease. J. Mol. Med. 2014, 92, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Polak-Jonkisz, D.; Purzyc, L. Ca2+ influx versus efflux during eryptosis in uremic erythrocytes. Blood Purif. 2012, 34, 209–210. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Salinas, J.V.; Munoz-Reyes, E.G.; Guerrero-Romero, J.F.; Rodriguez-Moran, M.; Bracho-Riquelme, R.L.; Carrera-Gracia, M.A.; Quintanar-Escorza, M.A. Eryptosis and oxidative damage in type 2 diabetic mellitus patients with chronic kidney disease. Mol. Cell. Biochem. 2011, 357, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.A.; Beringer, O.; Nicolay, J.P.; Amon, O.; Kempe, D.S.; Hermle, T.; Attanasio, P.; Akel, A.; Schafer, R.; Friedrich, B.; et al. Suicidal death of erythrocytes in recurrent hemolytic uremic syndrome. J. Mol. Med. 2006, 84, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Nicolay, J.P.; Schneider, J.; Niemoeller, O.M.; Artunc, F.; Portero-Otin, M.; Haik, G., Jr.; Thornalley, P.J.; Schleicher, E.; Wieder, T.; Lang, F. Stimulation of suicidal erythrocyte death by methylglyoxal. Cell. Physiol. Biochem. 2006, 18, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Lang, E.; Gatidis, S.; Freise, N.F.; Bock, H.; Kubitz, R.; Lauermann, C.; Orth, H.M.; Klindt, C.; Schuier, M.; Keitel, V.; et al. Conjugated bilirubin triggers anemia by inducing erythrocyte death. Hepatology 2015, 61, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Kempe, D.S.; Akel, A.; Lang, P.A.; Hermle, T.; Biswas, R.; Muresanu, J.; Friedrich, B.; Dreischer, P.; Wolz, C.; Schumacher, U.; et al. Suicidal erythrocyte death in sepsis. J. Mol. Med. 2007, 85, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.A.; Schenck, M.; Nicolay, J.P.; Becker, J.U.; Kempe, D.S.; Lupescu, A.; Koka, S.; Eisele, K.; Klarl, B.A.; Rubben, H.; et al. Liver cell death and anemia in wilson disease involve acid sphingomyelinase and ceramide. Nat. Med. 2007, 13, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Harrison, H.E.; Bunting, H.; Ordway, N.K.; Albrink, W.S. The pathogenesis of the renal injury produced in the dog by hemoglobin or methemoglobin. J. Exp. Med. 1947, 86, 339–356. [Google Scholar] [CrossRef] [PubMed]
- Kirk, K. Membrane transport in the malaria-infected erythrocyte. Physiol. Rev. 2001, 81, 495–537. [Google Scholar] [PubMed]
- Ayi, K.; Giribaldi, G.; Skorokhod, A.; Schwarzer, E.; Prendergast, P.T.; Arese, P. 16α-Bromoepiandrosterone, an antimalarial analogue of the hormone dehydroepiandrosterone, enhances phagocytosis of ring stage parasitized erythrocytes: A novel mechanism for antimalarial activity. Antimicrob. Agents Chemother. 2002, 46, 3180–3184. [Google Scholar] [CrossRef] [PubMed]
- Ayi, K.; Turrini, F.; Piga, A.; Arese, P. Enhanced phagocytosis of ring-parasitized mutant erythrocytes: A common mechanism that may explain protection against falciparum malaria in sickle trait and beta-thalassemia trait. Blood 2004, 104, 3364–3371. [Google Scholar] [CrossRef] [PubMed]
- Cappadoro, M.; Giribaldi, G.; O’Brien, E.; Turrini, F.; Mannu, F.; Ulliers, D.; Simula, G.; Luzzatto, L.; Arese, P. Early phagocytosis of glucose-6-phosphate dehydrogenase (G6PD)-deficient erythrocytes parasitized by plasmodium falciparum may explain malaria protection in G6PD deficiency. Blood 1998, 92, 2527–2534. [Google Scholar] [PubMed]
- Koka, S.; Huber, S.M.; Boini, K.M.; Lang, C.; Foller, M.; Lang, F. Lead decreases parasitemia and enhances survival of Plasmodium berghei-infected mice. Biochem. Biophys. Res. Commun. 2007, 363, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Koka, S.; Lang, C.; Niemoeller, O.M.; Boini, K.M.; Nicolay, J.P.; Huber, S.M.; Lang, F. Influence of no synthase inhibitor L-NAME on parasitemia and survival of Plasmodium berghei infected mice. Cell. Physiol. Biochem. 2008, 21, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Borst, O.; Abed, M.; Alesutan, I.; Towhid, S.T.; Qadri, S.M.; Foller, M.; Gawaz, M.; Lang, F. Dynamic adhesion of eryptotic erythrocytes to endothelial cells via CXCL16/SR-PSOX. Am. J. Physiol. Cell. Physiol. 2012, 302, C644–C651. [Google Scholar] [CrossRef] [PubMed]
- Andrews, D.A.; Low, P.S. Role of red blood cells in thrombosis. Curr. Opin. Hematol. 1999, 6, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.M.; Bae, O.N.; Lim, K.M.; Noh, J.Y.; Lee, M.Y.; Jung, Y.S.; Chung, J.H. Lysophosphatidic acid induces thrombogenic activity through phosphatidylserine exposure and procoagulant microvesicle generation in human erythrocytes. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Zwaal, R.F.; Comfurius, P.; Bevers, E.M. Surface exposure of phosphatidylserine in pathological cells. Cell. Mol. Life Sci. 2005, 62, 971–988. [Google Scholar] [CrossRef] [PubMed]
- Closse, C.; Dachary-Prigent, J.; Boisseau, M.R. Phosphatidylserine-related adhesion of human erythrocytes to vascular endothelium. Br. J. Haematol. 1999, 107, 300–302. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, P.G.; Chang, S.H.; Rettig, M.P.; Neely, J.E.; Hillery, C.A.; Smith, B.D.; Low, P.S. Altered erythrocyte endothelial adherence and membrane phospholipid asymmetry in hereditary hydrocytosis. Blood 2003, 101, 4625–4627. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, A.; Di Pietro, N.; Sirolli, V.; Giardinelli, A.; Di Silvestre, S.; Amoroso, L.; Di Tomo, P.; Capani, F.; Consoli, A.; Bonomini, M. Mechanisms of uremic erythrocyte-induced adhesion of human monocytes to cultured endothelial cells. J. Cell. Physiol. 2007, 213, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Wood, B.L.; Gibson, D.F.; Tait, J.F. Increased erythrocyte phosphatidylserine exposure in sickle cell disease: Flow-cytometric measurement and clinical associations. Blood 1996, 88, 1873–1880. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Officioso, A.; Manna, C.; Alzoubi, K.; Lang, F. Triggering of Erythrocyte Death by Triparanol. Toxins 2015, 7, 3359-3371. https://doi.org/10.3390/toxins7083359
Officioso A, Manna C, Alzoubi K, Lang F. Triggering of Erythrocyte Death by Triparanol. Toxins. 2015; 7(8):3359-3371. https://doi.org/10.3390/toxins7083359
Chicago/Turabian StyleOfficioso, Arbace, Caterina Manna, Kousi Alzoubi, and Florian Lang. 2015. "Triggering of Erythrocyte Death by Triparanol" Toxins 7, no. 8: 3359-3371. https://doi.org/10.3390/toxins7083359
APA StyleOfficioso, A., Manna, C., Alzoubi, K., & Lang, F. (2015). Triggering of Erythrocyte Death by Triparanol. Toxins, 7(8), 3359-3371. https://doi.org/10.3390/toxins7083359