
Metabolism of HT-2 Toxin and T-2 Toxin in Oats

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
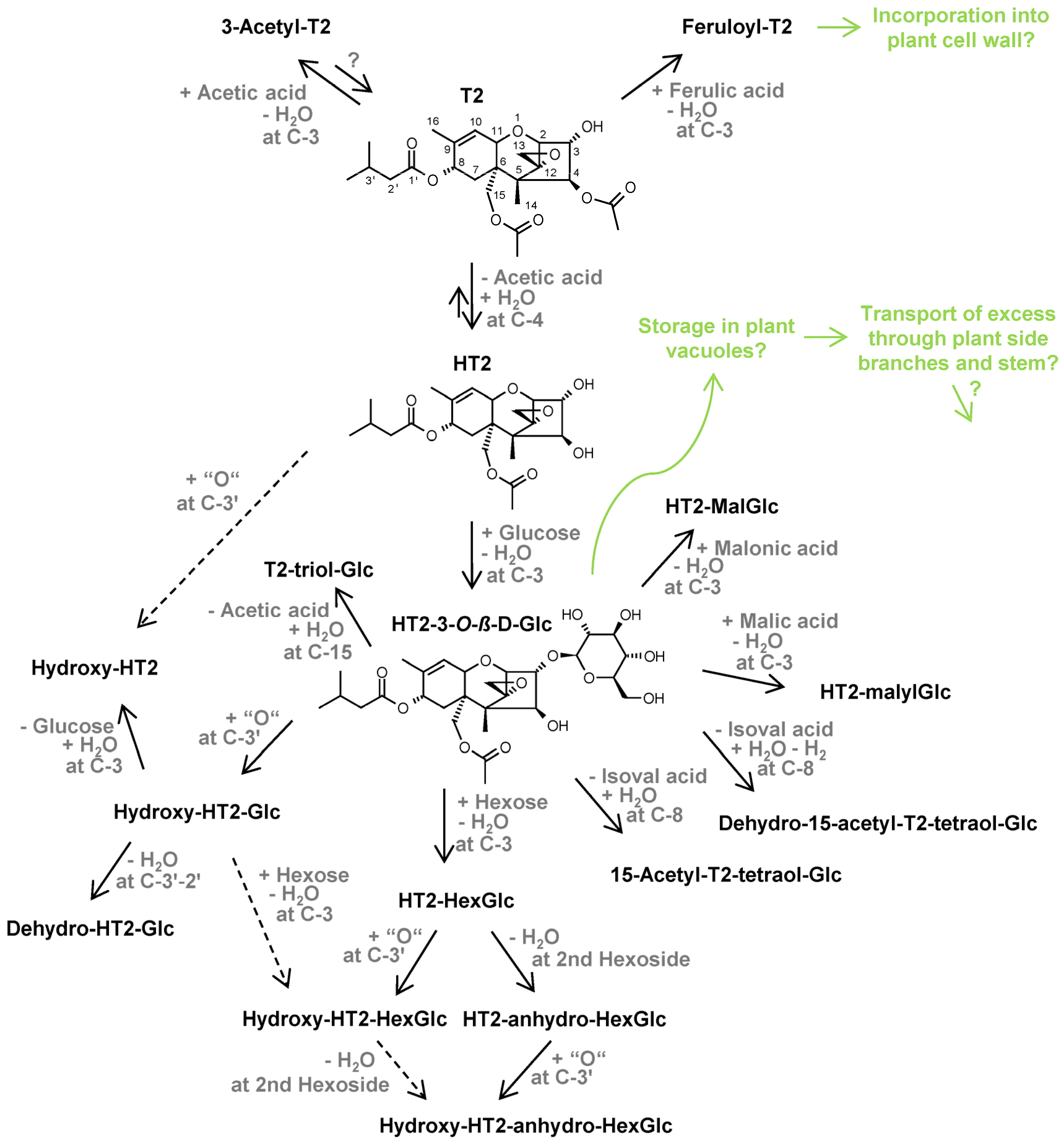
2.1. Overview of Annotated HT2 and T2 Metabolites in Oats
2.2. Structure Elucidation by LC-HRMS/MS
2.2.1. Confirmation of Previously Found HT2 and T2 Metabolites
2.2.2. Elucidation of Novel HT2 and T2 Metabolites
2.3. Kinetics of Metabolite Formation and Distribution
2.3.1. Absolute Quantification
2.3.2. Relative Quantification
2.3.3. Mobility of Parent Toxins and HT2-3-O-β-d-Glc in Oats
3. Conclusions
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Plant Cultivation
4.3. Treatment and Sampling of Oat Plants
4.3.1. Qualitative Screening Experiment
4.3.2. Time Course Experiment
4.4. Sample Preparation
4.4.1. Qualitative Screening Experiment
4.4.2. Time Course Experiment
4.5. Analysis by LC-HRMS and LC-HRMS/MS
4.5.1. Qualitative Screening Experiment
4.5.2. Structure Annotation and Quantification (Time Course Experiment)
4.6. Metabolite Recognition by MetExtract II (Module TracExtract)
4.7. Annotation of Unknown Metabolites by MetExtract II (Module FragExtract)
4.8. Quantification Experiments
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Edwards, S.G.; Barrier-Guillot, B.; Clasen, P.-E.; Hietaniemi, V.; Pettersson, H. Emerging issues of HT-2 and T-2 toxins in European cereal production. World Mycotoxin J. 2009, 2, 173–179. [Google Scholar] [CrossRef]
- Lemmens, M.; Steiner, B.; Sulyok, M.; Nicholson, P.; Mesterhazy, A.; Buerstmayr, H. Masked mycotoxins: Does breeding for enhanced Fusarium head blight resistance result in more deoxynivalenol-3-glucoside in new wheat varieties? World Mycotoxin J. 2016. [Google Scholar] [CrossRef]
- Gottschalk, C.; Barthel, J.; Engelhardt, G.; Bauer, J.; Meyer, K. Simultaneous determination of type A, B and D trichothecenes and their occurrence in cereals and cereal products. Food Addit. Contam. 2009, 26, 1273–1289. [Google Scholar] [CrossRef]
- Kirinčič, S.; Škrjanc, B.; Kos, N.; Kozolc, B.; Pirnat, N.; Tavčar-Kalcher, G. Mycotoxins in cereals and cereal products in Slovenia—Official control of foods in the years 2008–2012. Food Control 2015, 50, 157–165. [Google Scholar] [CrossRef]
- Marin, S.; Ramos, A.J.; Cano-Sancho, G.; Sanchis, V. Mycotoxins: Occurrence, toxicology, and exposure assessment. Food Chem. Toxicol. 2013, 60, 218–237. [Google Scholar] [CrossRef] [PubMed]
- Krska, R.; Malachova, A.; Berthiller, F.; van Egmond, H.P. Determination of T-2 and HT-2 toxins in food and feed: An update. World Mycotoxin J. 2014, 7, 131–142. [Google Scholar] [CrossRef]
- European Food Safety Authority (EFSA) Panel on Contaminants in the Food Chain (CONTAM). Scientific Opinion on the risks for animal and public health related to the presence of T-2 and HT-2 toxin in food and feed. EFSA J. 2011, 9. [Google Scholar] [CrossRef]
- Commission Recommendation 2013/165/EU of 27 March 2013 on the presence of T-2 and HT-2 toxin in cereals and cereal products. Off. J. Eur. Commun. 2013, L91, 12–15.
- Cole, D.J. Detoxification and Activation of Agrochemicals in Plants. Pestic. Sci. 1994, 42, 209–222. [Google Scholar] [CrossRef]
- Coleman, J.O.D.; Blake-Kalff, M.M.A.; Davies, T.G.E. Detoxification of xenobiotics by plants: Chemical modification and vacuolar compartmentation. Trends Plant Sci. 1997, 2, 144–151. [Google Scholar] [CrossRef]
- Gougler, J.A.; Geiger, D.R. Uptake and Distribution of N-Phosphonomethylglycine in Sugar Beet Plants. Plant Physiol. 1981, 68, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Schröder, P.; Scheer, C.E.; Diekmann, F.; Stampfl, A. How Plants Cope with Foreign Compounds. Translocation of xenobiotic glutathione conjugates in roots of barley (Hordeum vulgare). Environ. Sci. Pollut. Res. 2007, 14, 114–122. [Google Scholar]
- Wu, Q.; Dohnal, V.; Kuča, K.; Yuan, Z. Trichothecenes: Structure-Toxic Activity Relationships. Curr. Drug Metab. 2013, 14, 641–660. [Google Scholar] [CrossRef] [PubMed]
- Berthiller, F.; Werner, U.; Sulyok, M.; Krska, R.; Hauser, M.-T.; Schuhmacher, R. Liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) determination of phase II metabolites of the mycotoxin zearalenone in the model plant Arabidopsis thaliana. Food Addit. Contam. 2006, 23, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Kluger, B.; Bueschl, C.; Lemmens, M.; Michlmayr, H.; Malachova, A.; Koutnik, A.; Maloku, I.; Berthiller, F.; Adam, G.; Krska, R.; et al. Biotransformation of the Mycotoxin Deoxynivalenol in Fusarium Resistant and Susceptible Near Isogenic Wheat Lines. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Pierron, A.; Mimoun, S.; Murate, L.S.; Loiseau, N.; Lippi, Y.; Bracarense, A.-P.F.L.; Liaubet, L.; Schatzmayr, G.; Berthiller, F.; Moll, W.-D.; et al. Intestinal toxicity of the masked mycotoxin deoxynivalenol-3-β-d-glucoside. Arch. Toxicol. 2016, 90, 2037–2046. [Google Scholar] [CrossRef] [PubMed]
- Gareis, M.; Bauer, J.; Thiem, J.; Plank, G.; Grabley, S.; Gedek, B. Cleavage of zearalenone glycoside, a ‘masked’ mycotoxin during digestion in swine. J. Vet. Med. B 1990, 37, 236–240. [Google Scholar] [CrossRef]
- Berthiller, F.; Brera, C.; Crews, C.; Iha, M.H.; Krska, R.; Lattanzio, V.M.T.; MacDonald, S.; Malone, R.J.; Maragos, C.; Solfrizzo, M.; et al. Developments in mycotoxin analysis: An update for 2014–2015. World Mycotoxin J. 2016, 9, 5–29. [Google Scholar] [CrossRef]
- Berthiller, F.; Crews, C.; Dall’Asta, C.; De Saeger, S.; Haesaert, G.; Karlovsky, P.; Oswald, I.P.; Seefelder, W.; Speijers, G.; Stroka, J. Masked mycotoxins: A review. Mol. Nutr. Food Res. 2013, 57, 165–186. [Google Scholar] [CrossRef] [PubMed]
- Sulyok, M.; Krska, R.; Schuhmacher, R. Application of an LC-MS/MS based multi-mycotoxin method for the semi-quantitative determination of mycotoxins occurring in different types of food infected by moulds. Food Chem. 2010, 119, 408–416. [Google Scholar] [CrossRef]
- Sulyok, M.; Beed, F.; Boni, S.; Abass, A.; Mukunzi, A.; Krska, R. Quantitation of multiple mycotoxins and cyanogenic glucosides in cassava samples from Tanzania and Rwanda by an LC-MS/MS-based multi-toxin method. Food Addit. Contam. Part A 2015, 32, 488–502. [Google Scholar] [CrossRef] [PubMed]
- Lattanzio, V.M.T.; Ciasca, B.; Powers, S.; Visconti, A. Improved method for the simultaneous determination of aflatoxins, ochratoxin A and Fusarium toxins in cereals and derived products by liquid chromatography-tandem mass spectrometry after multi-toxin immunoaffinity clean up. J. Chromatogr. A 2014, 1354, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Bueschl, C.; Krska, R.; Kluger, B.; Schuhmacher, R. Isotopic labeling-assisted metabolomics using LC-MS. Anal. Bioanal. Chem. 2013, 405, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Kluger, B.; Bueschl, C.; Lemmens, M.; Berthiller, F.; Häubl, G.; Jaunecker, G.; Adam, G.; Krska, R.; Schuhmacher, R. Stable isotopic labelling-assisted untargeted metabolic profiling reveals novel conjugates of the mycotoxin deoxynivalenol in wheat. Anal. Bioanal. Chem. 2013, 405, 5031–5036. [Google Scholar] [CrossRef] [PubMed]
- Kluger, B.; Bueschl, C.; Neumann, N.; Stückler, R.; Doppler, M.; Chassy, A.W.; Waterhouse, A.L.; Rechthaler, J.; Kampleitner, N.; Thallinger, G.G.; et al. Untargeted Profiling of Tracer-Derived Metabolites Using Stable Isotopic Labeling and Fast Polarity-Switching LC−ESI-HRMS. Anal. Chem. 2014, 86, 11533–11537. [Google Scholar] [CrossRef] [PubMed]
- Lattanzio, V.M.T.; Visconti, A.; Haidukowski, M.; Pascale, M. Identification and characterization of new Fusarium masked mycotoxins, T2 and HT2 glycosyl derivatives, in naturally contaminated wheat and oats by liquid chromatography-high-resolution mass spectrometry. J. Mass Spectrom. 2012, 47, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Busman, M.; Poling, S.M.; Maragos, C.M. Observation of T-2 Toxin and HT-2 Toxin Glucosides from Fusarium sporotrichioides by Liquid Chromatography Coupled to Tandem Mass Spectrometry (LC-MS/MS). Toxins 2011, 3, 1554–1568. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, E.; Monaci, L.; Pascale, M.; Visconti, A. Fate of deoxynivalenol, T-2 and HT-2 toxins and their glucoside conjugates from flour to bread: An investigation by high-performance liquid chromatography high-resolution mass spectrometry. Food Addit. Contam. Part A 2013, 30, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Sakamoto, S.; Sago, Y.; Nagashima, H. Detection of Type A Trichothecene Di-Glucosides Produced in Corn by High-Resolution Liquid Chromatography-Orbitrap Mass Spectrometry. Toxins 2013, 5, 590–604. [Google Scholar] [CrossRef] [PubMed]
- Veprikova, Z.; Vaclavikova, M.; Lacina, O.; Dzuman, Z.; Zachariasova, M.; Hajslova, J. Occurrence of mono- and di-glycosylated conjugates of T-2 and HT-2 toxins in naturally contaminated cereals. World Mycotoxin J. 2012, 5, 231–240. [Google Scholar] [CrossRef]
- McCormick, S.P.; Kato, T.; Maragos, C.M.; Busman, M.; Lattanzio, V.M.T.; Galaverna, G.; Dall-Asta, C.; Crich, D.; Price, N.P.J.; Kurtzman, C.P. Anomericity of T-2 Toxin-glucoside: Masked Mycotoxin in Cereal Crops. J. Agric. Food Chem. 2015, 63, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Meng-Reiterer, J.; Varga, E.; Nathanail, A.V.; Bueschl, C.; Rechthaler, J.; McCormick, S.P.; Michlmayr, H.; Malachová, A.; Fruhmann, P.; Adam, G.; Berthiller, F.; Lemmens, M.; Schuhmacher, R. Tracing the metabolism of HT-2 toxin and T-2 toxin in barley by isotope-assisted untargeted screening and quantitative LC-HRMS analysis. Anal. Bioanal. Chem. 2015, 407, 8019–8033. [Google Scholar] [CrossRef] [PubMed]
- Nathanail, A.V.; Varga, E.; Meng-Reiterer, J.; Bueschl, C.; Michlmayr, H.; Malachova, A.; Fruhmann, P.; Jestoi, M.N.; Peltonen, K.; Adam, G.; Lemmens, M.; Schuhmacher, R.; Berthiller, F. Metabolism of the Fusarium Mycotoxins T‑2 Toxin and HT‑2 Toxin in Wheat. J. Agric. Food Chem. 2015, 63, 7862–7872. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, G.; Ubukata, T.; Nozue, H.; Kobayashi, Y.; Takahi, M.; Yamamoto, H.; Hayashida, N. Malonylation is a key reaction in the metabolism of xenobiotic phenolic glucosides in Arabidopsis and tobacco. Plant J. 2010, 63, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Bockers, M.; Rivero, C.; Thiede, B.; Jankowski, T.; Schmidt, B. Uptake, Translocation, and Metabolism of 3,4-Dichloroaniline in Soybean and Wheat Plants. Z. Naturforsch. 1994, 49c, 719–726. [Google Scholar]
- Abdel-Farid, I.B.; Kim, H.K.; Choi, Y.H.; Verpoorte, R. Metabolic Characterization of Brassica rapa Leaves by NMR Spectroscopy. J. Agric. Food Chem. 2007, 55, 7936–7943. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Matoba, Y.; Katagi, T. Application of Separated Leaf Cell Suspension to Xenobiotic Metabolism in Plant. J. Agric. Food Chem. 2009, 57, 6982–6989. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Library of Medicine. National Center for Biotechnology Information. PUBCHEM Database. Compound Summary for CID 525. MS-MS-Spectra of NIST Number 1118542 and 1052306. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/malic_acid#section=MS-MS (accessed on 16 August 2016).
- Hedin, P.A.; Phillips, V.A. Chemical Ionization (Methane) Mass Spectrometry of Sugars and Their Derivatives. J. Agric. Food Chem. 1991, 39, 1106–1109. [Google Scholar] [CrossRef]
- Iiyama, K.; Lam, T.B.-T.; Stone, B.A. Covalent Cross-Links in the Cell Wall. Plant Physiol. 1994, 104, 315–320. [Google Scholar] [CrossRef] [PubMed]
- McKeehen, J.D.; Busch, R.H.; Fulcher, R.G. Evaluation of Wheat (Triticum aestivum L.) Phenolic Acids during Grain Development and Their Contribution to Fusarium Resistance. J. Agric. Food Chem. 1999, 47, 1476–1482. [Google Scholar] [CrossRef] [PubMed]
- Carpaneto, A.; Geiger, D.; Bamberg, E.; Sauer, N.; Fromm, J.; Hedrich, R. Phloem-localized, proton-coupled sucrose carrier ZmSUT1 mediates sucrose efflux under the control of the sucrose gradient and the proton motive force. J. Biol. Chem. 2005, 280, 21437–21443. [Google Scholar] [CrossRef] [PubMed]
- Saatzucht Edelhof. Available online: http://www.saatzucht.edelhof.at/en/ (accessed on 28 November 2016).
- Kessner, D.; Chambers, M.; Burke, R.; Agusand, D.; Mallick, P. ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics 2008, 24, 2534–2536. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Kibbe, W.A.; Lin, S.M. Improved peak detection in mass spectrum by incorporating continuous wavelet transform-based pattern matching. Bioinformatics 2006, 22, 2059–2065. [Google Scholar] [CrossRef] [PubMed]
- Neumann, N.K.; Lehner, S.M.; Kluger, B.; Bueschl, C.; Sedelmaier, K.; Lemmens, M.; Krska, R.; Schuhmacher, R. Automated LC-HRMS(/MS) approach for the annotation of fragment ions derived from stable isotope labeling-assisted untargeted metabolomics. Anal. Chem. 2014, 86, 7320–7327. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Fiehn, O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinform. 2007, 8, 105–124. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Metabolite | Retention Time (min) | Elemental Composition a | Accurate Mass b | Adduct b | nc c | Mass Error (ppm) |
|---|---|---|---|---|---|---|---|
| HT2 * | 15.69 | C22H32O8 | 442.2446 | [M+NH4]+ | 22 | 2.4 | |
| 1 | 15-Acetyl-T2-tetraol-Glc ** | 5.45 | C23H34O12 | 520.2408 | [M+NH4]+ | 17 | 3.7 |
| 2 | Dehydro-15-acetyl-T2-tetraol-Glc d,*** | 8.09 | C23H32O12 | 518.2250 | [M+NH4]+ | 17 | 3.5 |
| 3 | Hydroxy-HT2-HexGlc ** | 10.21 | C34H52O19 | 782.3468 | [M+NH4]+ | 22 | 3.4 |
| 4 | Hydroxy-HT2-HexGlc ** | 10.88 | C34H52O19 | 809.3100 | [M+HCOO]− | 22 | 1.9 |
| 5 | Hydroxy-HT2-Glc ** | 11.08 | C28H42O14 | 620.2927 | [M+NH4]+ | 22 | 2.3 |
| 6 | Hydroxy-HT2 ** | 11.71 | C22H32O9 | 458.2398 | [M+NH4]+ | 22 | 2.9 |
| 7 | Hydroxy-HT2-anhydro-HexGlc ** | 12.37 | C34H50O18 | 764.3355 | [M+NH4]+ | 22 | 2.6 |
| 8 | T2-triol-Glc ** | 13.81 | C26H40O12 | 589.2508 | [M+HCOO]− | 20 | 1.1 |
| 9 | Dehydro-HT2-Glc ** | 14.05 | C28H40O13 | 629.2458 | [M+HCOO]− | 22 | 1.1 |
| 10 | HT2-HexGlc ** | 14.23 | C34H52O18 | 766.3509 | [M+NH4]+ | 22 | 2.2 |
| 11 | HT2-HexGlc ** | 14.76 | C34H52O18 | 766.3511 | [M+NH4]+ | 22 | 2.5 |
| 12 | HT2-malylGlc ** | 14.85 | C32H46O17 | 720.3087 | [M+NH4]+ | 22 | 1.9 |
| 13 | HT2-3-O-β-d-Glc * | 15.00 | C28H42O13 | 604.2973 | [M+NH4]+ | 22 | 1.5 |
| 14 | HT2-MalGlc d,** | 15.11 | C31H44O16 | 690.2994 | [M+NH4]+ | 22 | 3.8 |
| 15 | HT2-anhydro-HexGlc ** | 15.51 | C34H50O17 | 748.3404 | [M+NH4]+ | 22 | 2.4 |
| 16 | T2 d,* | 16.87 | C24H34O9 | 484.2549 | [M+NH4]+ | 22 | 1.6 |
| ID | Metabolite | Retention Time (min) | Elemental Composition a | Accurate Mass b | Adduct b | nc c | Mass Error (ppm) |
|---|---|---|---|---|---|---|---|
| T2 * | 16.83 | C24H34O9 | 484.2548 | [M+NH4]+ | 24 | 1.4 | |
| 1 | 15-Acetyl-T2-tetraol-Glc d,** | 5.46 | C23H34O12 | 520.2407 | [M+NH4]+ | 17 | 3.6 |
| 2 | Dehydro-15-acetyl-T2-tetraol-Glc d,*** | 8.10 | C23H32O12 | 518.2248 | [M+NH4]+ | 17 | 3.1 |
| 3 | Hydroxy-HT2-HexGlc d,** | 10.23 | C34H52O19 | 782.3462 | [M+NH4]+ | 22 | 2.7 |
| 5 | Hydroxy-HT2-Glc ** | 11.08 | C28H42O14 | 620.2926 | [M+NH4]+ | 22 | 2.1 |
| 6 | Hydroxy-HT2 ** | 11.71 | C22H32O9 | 458.2396 | [M+NH4]+ | 22 | 2.5 |
| 7 | Hydroxy-HT2-anhydro-HexGlc d,** | 12.36 | C34H50O18 | 764.3348 | [M+NH4]+ | 22 | 1.7 |
| 8 | T2-triol-Glc ** | 13.81 | C26H40O12 | 589.2508 | [M+HCOO]− | 20 | 1.1 |
| 9 | Dehydro-HT2-Glc ** | 14.04 | C28H40O13 | 629.2458 | [M+HCOO]− | 22 | 1.1 |
| 10 | HT2-HexGlc ** | 14.23 | C34H52O18 | 766.3509 | [M+NH4]+ | 22 | 2.2 |
| 11 | HT2-HexGlc ** | 14.78 | C34H52O18 | 766.3511 | [M+NH4]+ | 22 | 2.5 |
| 12 | HT2-malylGlc ** | 14.84 | C32H46O17 | 720.3088 | [M+NH4]+ | 22 | 2.0 |
| 13 | HT2-3-O-β-d-Glc * | 14.99 | C28H42O13 | 604.2975 | [M+NH4]+ | 22 | 1.9 |
| 14 | HT2-MalGlc d,** | 15.11 | C31H44O16 | 690.2990 | [M+NH4]+ | 22 | 3.2 |
| 15 | HT2-anhydro-HexGlc ** | 15.50 | C34H50O17 | 729.2983 | [M−H]− | 22 | 1.1 |
| 17 | HT2 * | 15.68 | C22H32O8 | 442.2446 | [M+NH4]+ | 22 | 2.4 |
| 18 | 3-Acetyl-T2 * | 18.07 | C26H36O10 | 526.2653 | [M+NH4]+ | 24 | 1.2 |
| 19 | Feruloyl-T2 d,** | 19.17 | C34H42O12 | 660.3022 | [M+NH4]+ | 24 | 1.1 |
| Time Points (Days) | % of HT2 at 0 Days a Stem plus Side Branches | % of HT2 at 0 Days Non-Treated Spikelets |
|---|---|---|
| 0 | 0.0 ± 0.0 | <LOD |
| 1 | 4.4 ± 2.6 | <LOD |
| 3 | 3.0 ± 2.6 | <LOD |
| 7 | 2.4 ± 2.2 | <LOD |
| full-ripening | 1.2 ± 1.2 | <LOD |
| Time Points (Days) | % of T2 + HT2 at 0 Days a Stem plus Side Branches | % of T2 + HT2 at 0 Days Non-Treated Spikelets |
|---|---|---|
| 0 | 0.0 ± 0.0 | <LOD |
| 1 | 1.3 ± 0.9 | <LOD |
| 3 | 0.7 ± 0.3 | <LOD |
| 7 | 1.0 ± 0.8 | <LOD |
| full-ripening | 0.3 ± 0.3 | <LOD |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng-Reiterer, J.; Bueschl, C.; Rechthaler, J.; Berthiller, F.; Lemmens, M.; Schuhmacher, R. Metabolism of HT-2 Toxin and T-2 Toxin in Oats. Toxins 2016, 8, 364. https://doi.org/10.3390/toxins8120364
Meng-Reiterer J, Bueschl C, Rechthaler J, Berthiller F, Lemmens M, Schuhmacher R. Metabolism of HT-2 Toxin and T-2 Toxin in Oats. Toxins. 2016; 8(12):364. https://doi.org/10.3390/toxins8120364
Chicago/Turabian StyleMeng-Reiterer, Jacqueline, Christoph Bueschl, Justyna Rechthaler, Franz Berthiller, Marc Lemmens, and Rainer Schuhmacher. 2016. "Metabolism of HT-2 Toxin and T-2 Toxin in Oats" Toxins 8, no. 12: 364. https://doi.org/10.3390/toxins8120364
APA StyleMeng-Reiterer, J., Bueschl, C., Rechthaler, J., Berthiller, F., Lemmens, M., & Schuhmacher, R. (2016). Metabolism of HT-2 Toxin and T-2 Toxin in Oats. Toxins, 8(12), 364. https://doi.org/10.3390/toxins8120364