
QuEChERS Purification Combined with Ultrahigh-Performance Liquid Chromatography Tandem Mass Spectrometry for Simultaneous Quantification of 25 Mycotoxins in Cereals

Abstract
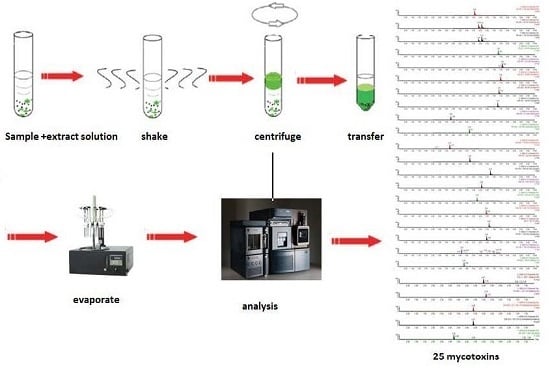
:
1. Introduction
2. Results and Discussion
2.1. Optimization of the Type of the Mobile Phase
2.2. Optimization of the Gradient Elution
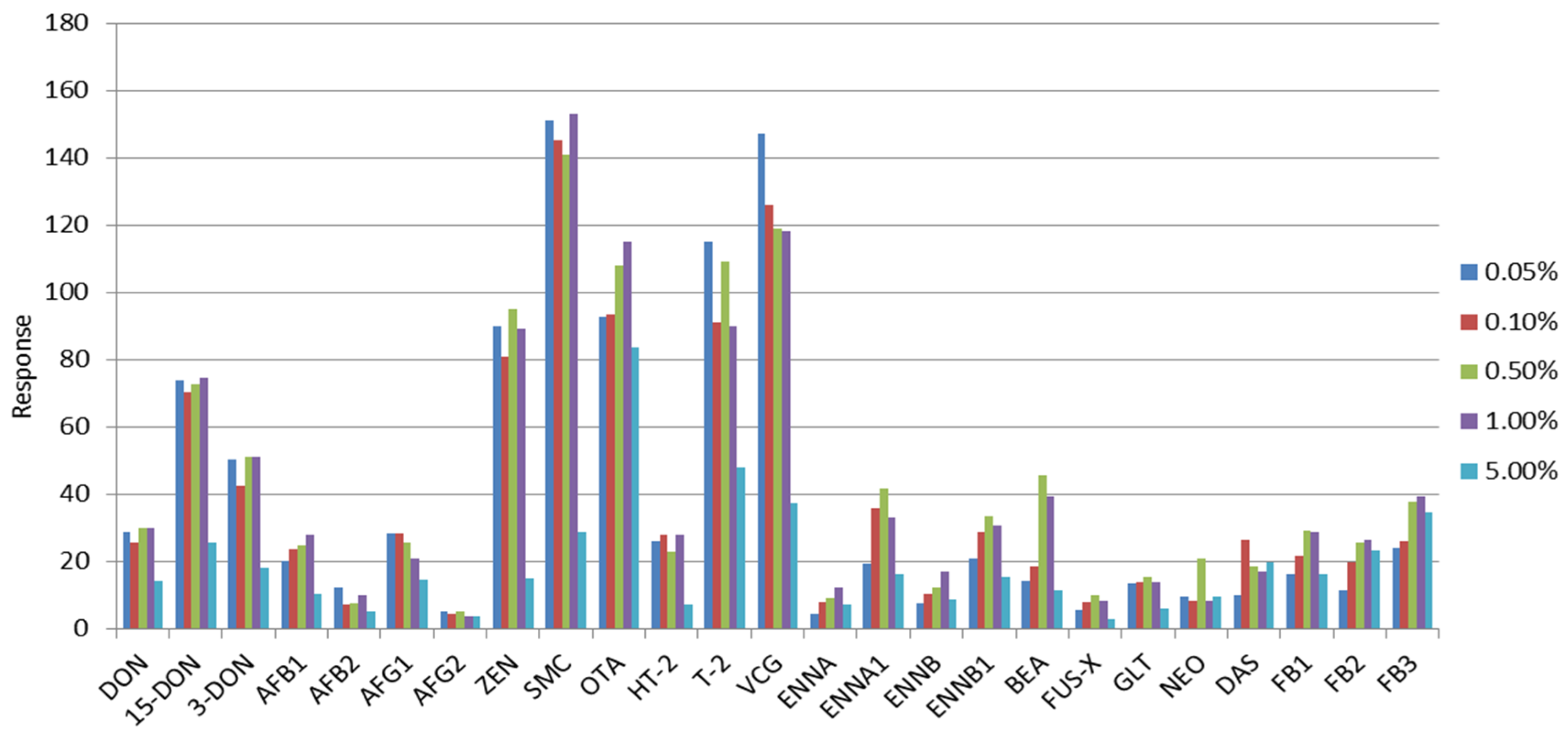
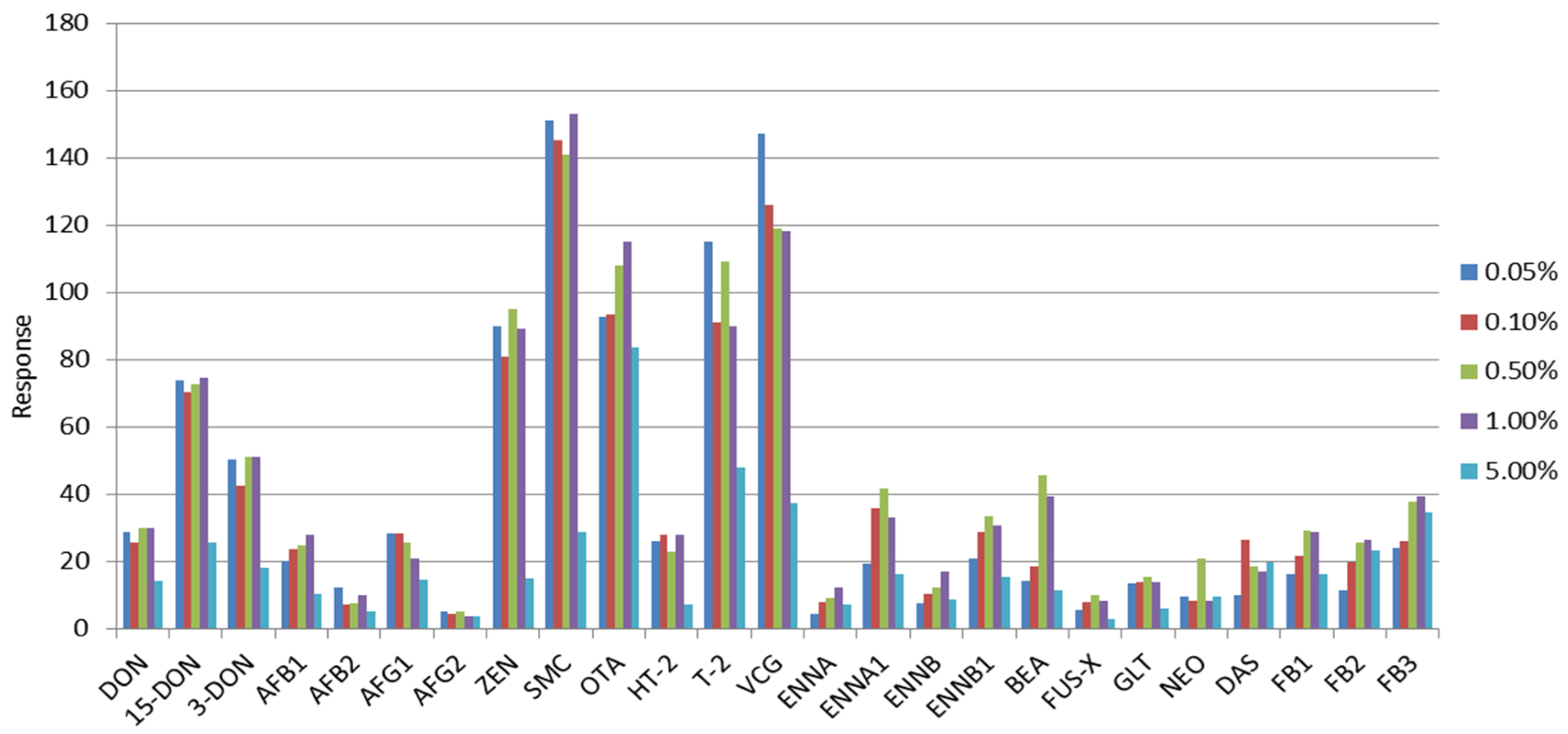
2.3. Selection of the Extraction Solvent and Purification Evaluation
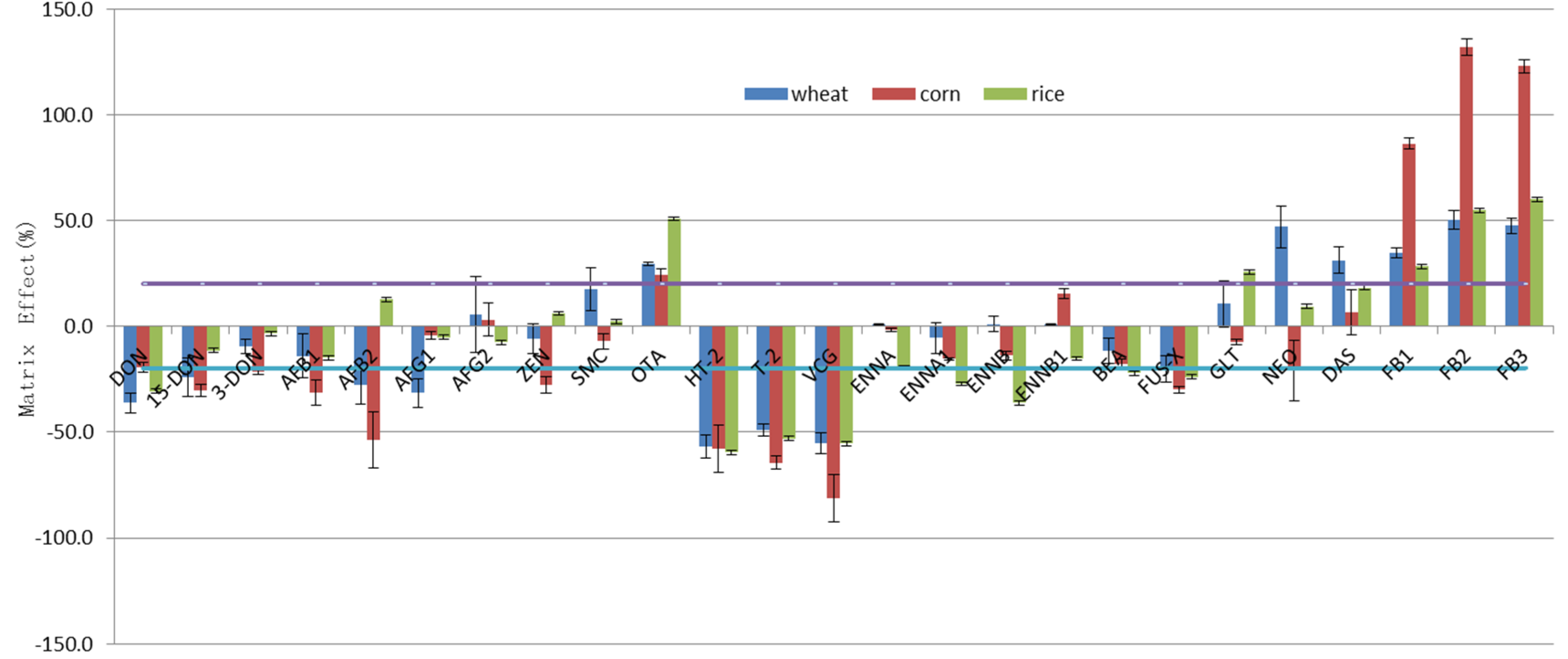
2.4. Evaluation of the Matrix Effect
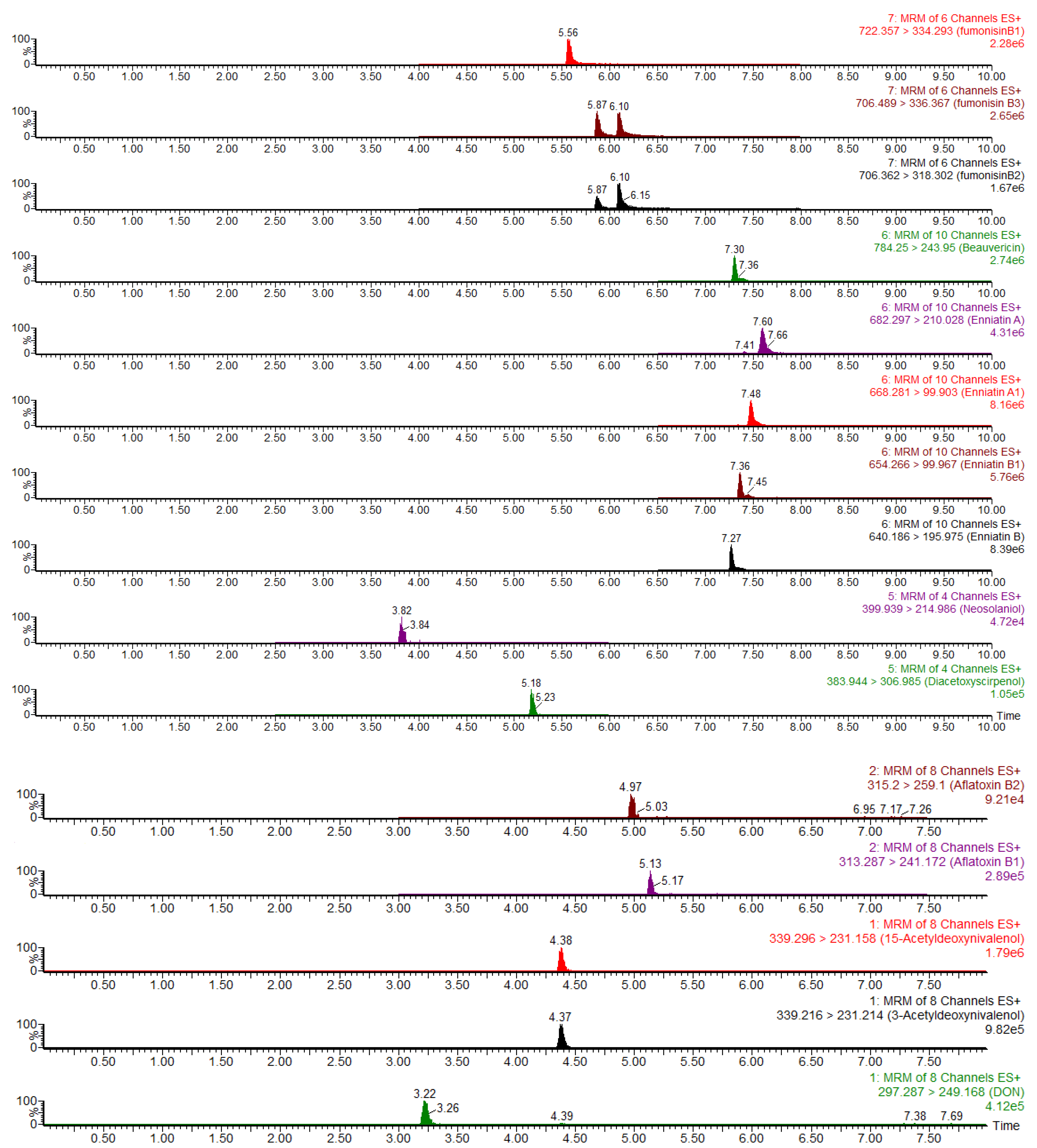
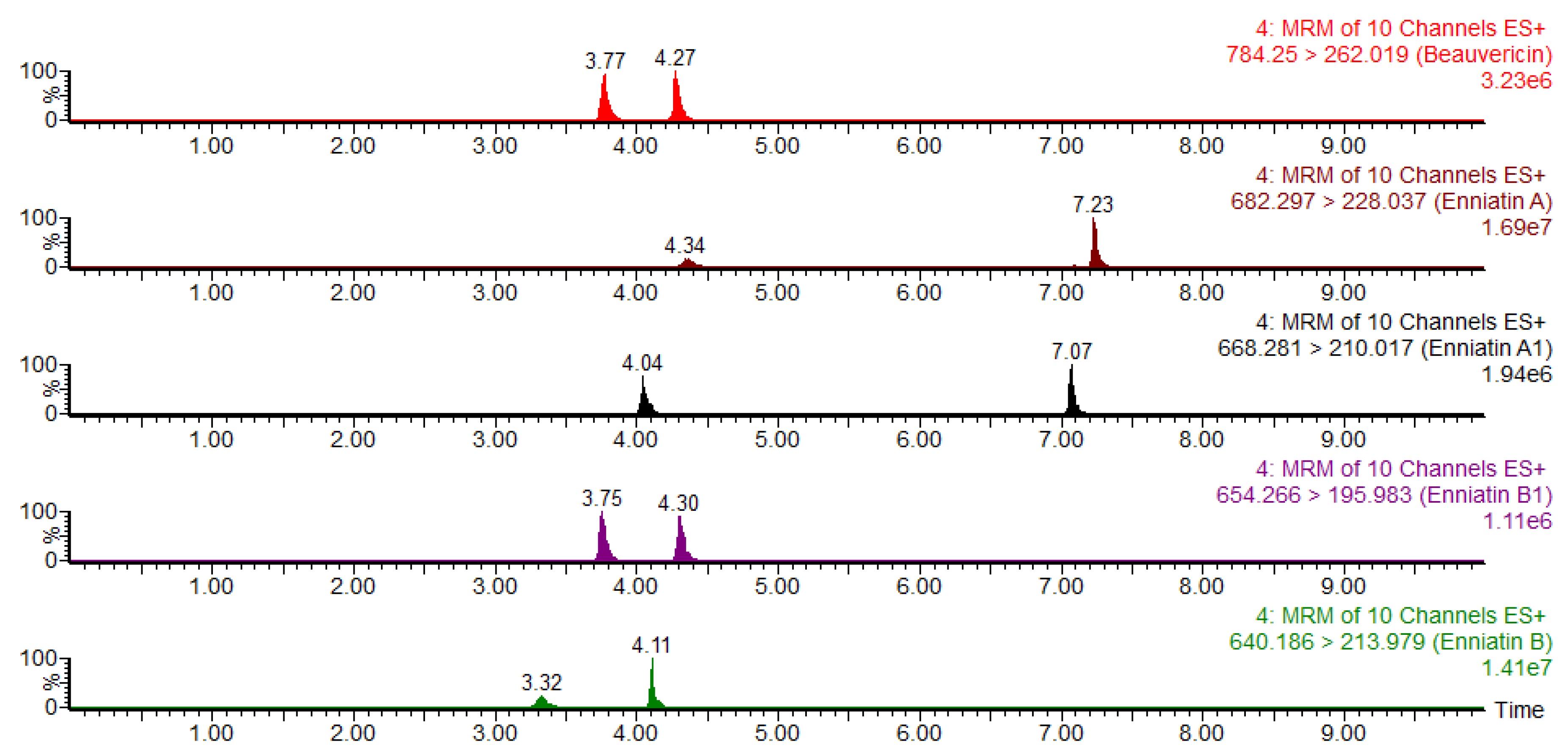
2.5. Method Validation
2.5.1. Calibration Curves and Linearity
2.5.2. Limit of Detection and Quantification
2.5.3. Method Accuracy and Precision
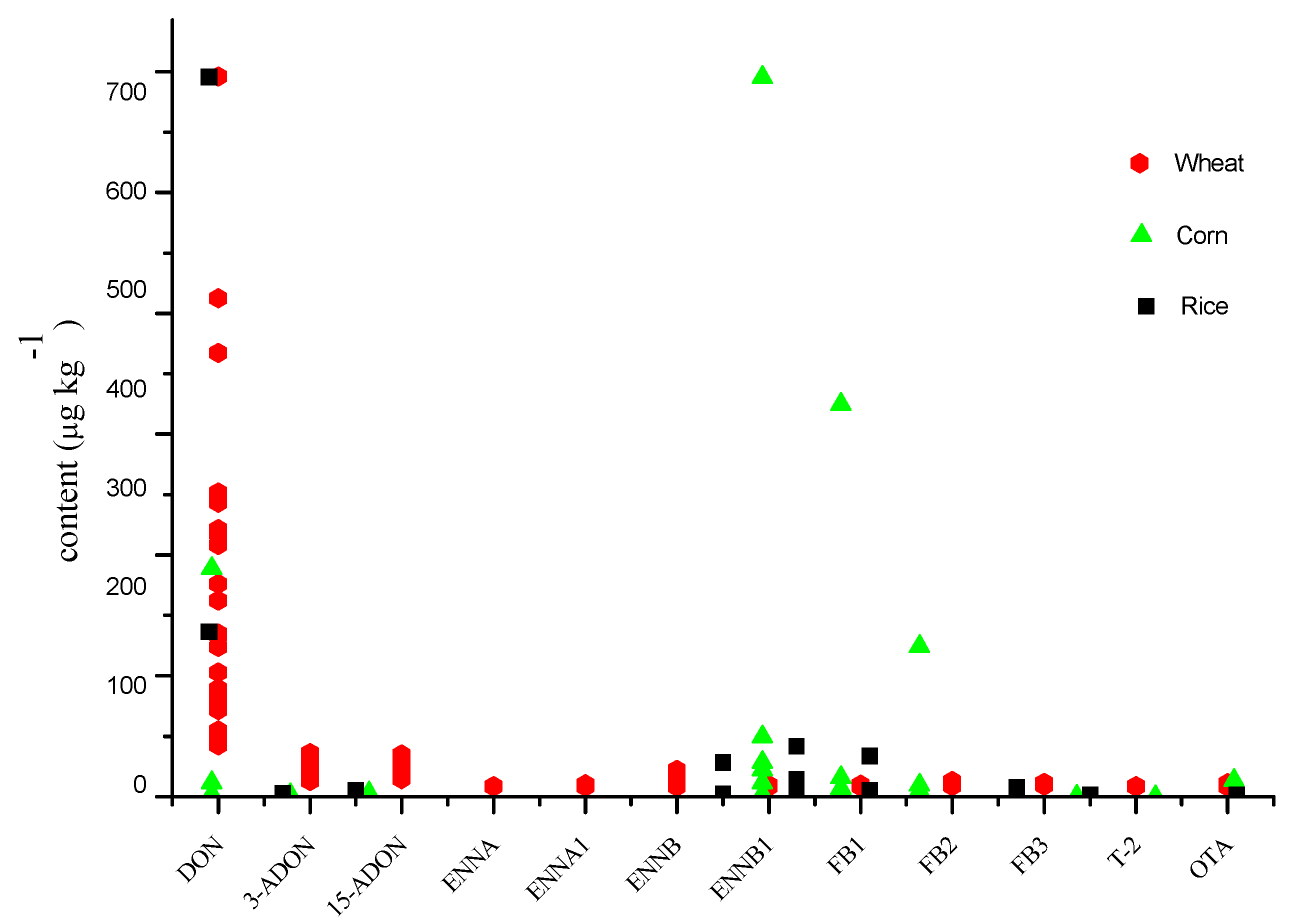
2.6. Application of the Developed Method on Real Samples
3. Conclusions
4. Experimental Section
4.1. Materials
4.2. Preparation of the Standard Solutions
4.3. Sample Preparation
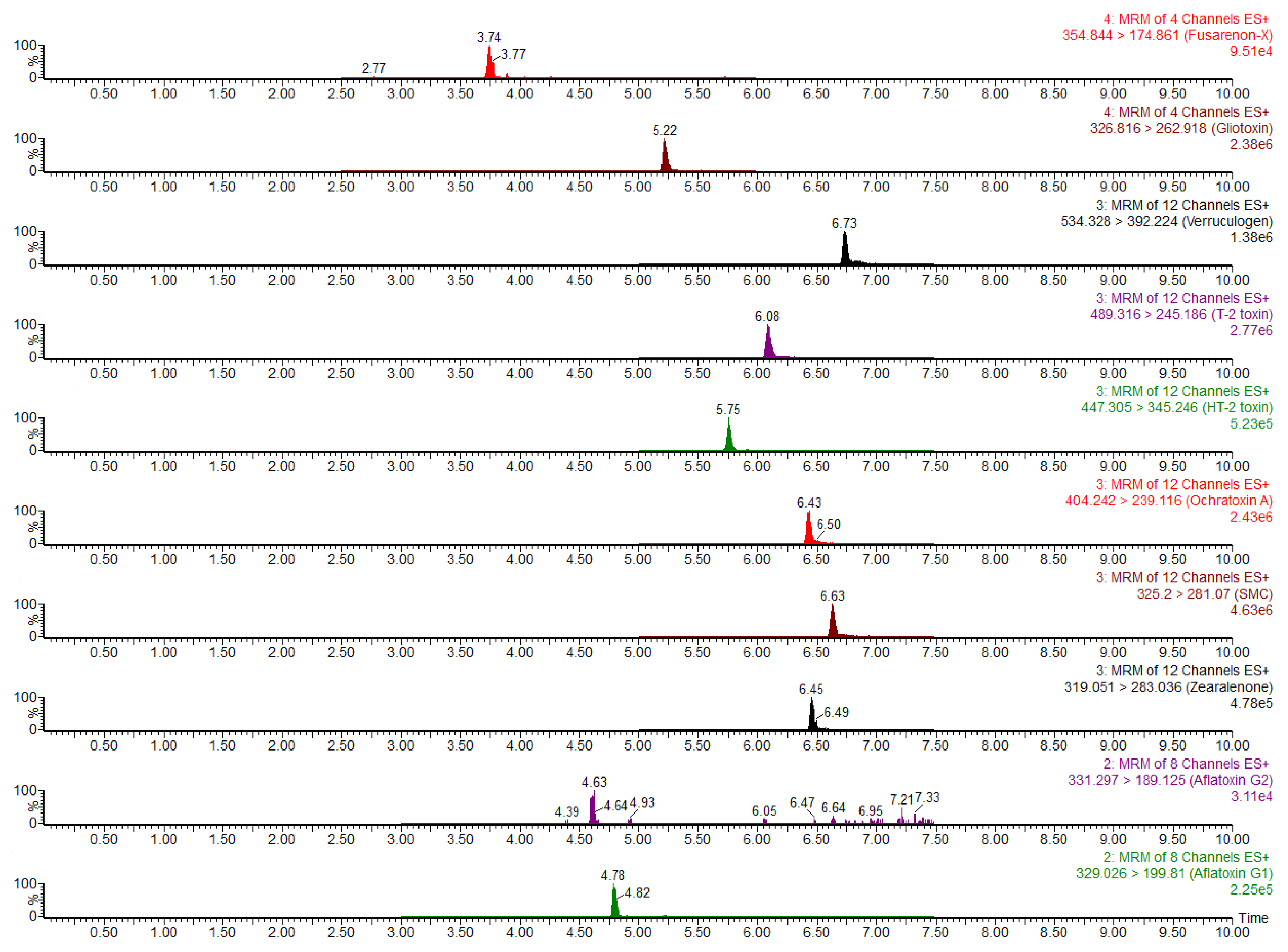
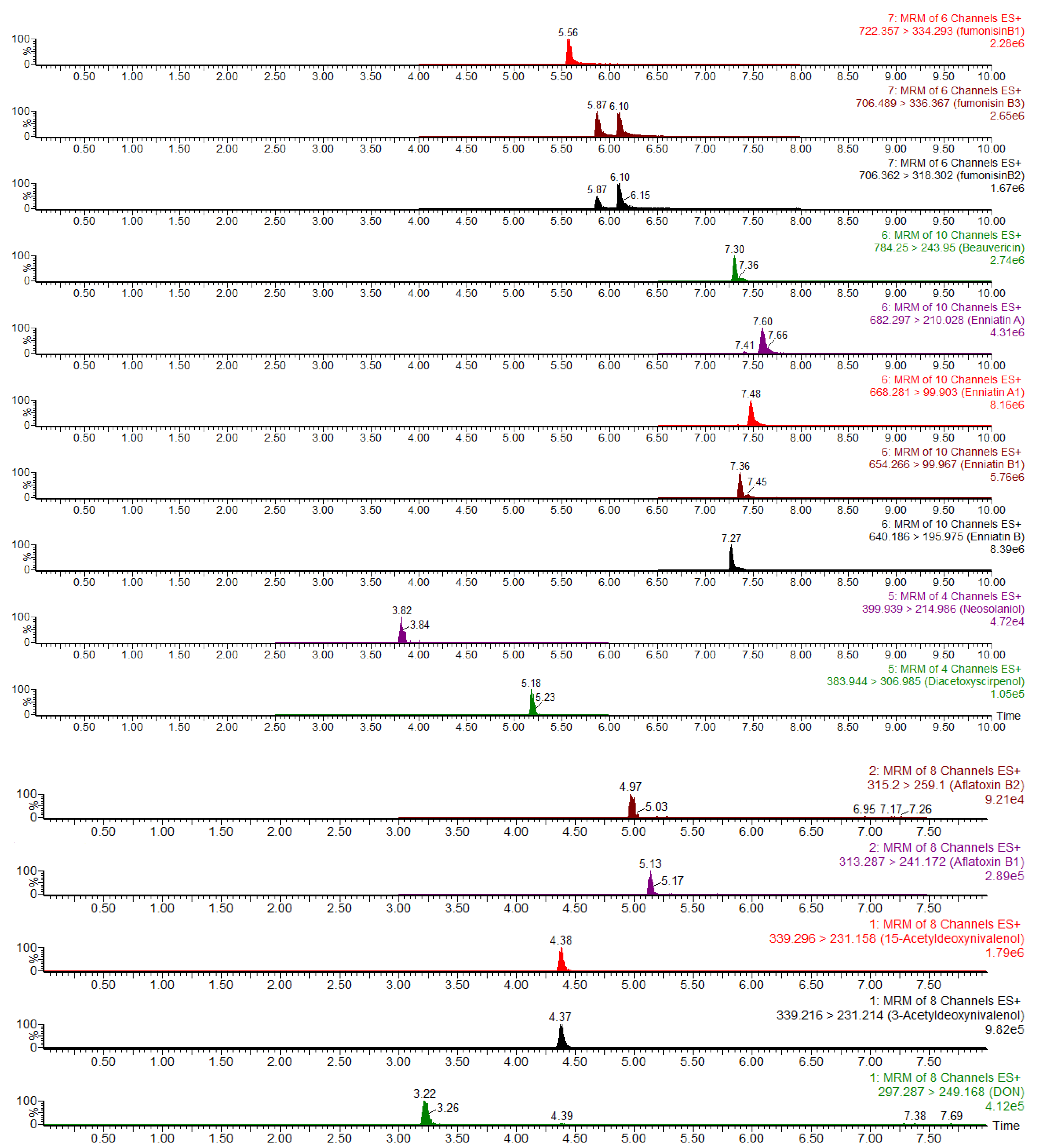
4.4. UPLC–MS/MS Conditions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Saeger, S.; Sibanda, L.; Desmet, A.; Van Peteghem, C. A collaborative study to validate novel field immunoassay kits for rapid mycotoxin detection. Int. J. Food Microbiol. 2002, 75, 135–142. [Google Scholar] [CrossRef]
- Tamura, M.; Mochizuki, N. Development of a Multi-mycotoxin Analysis in Beer-based Drinks by a Modified QuEChERS Method and Ultra-High-Performance Liquid Chromatography Coupled with Tandem Mass Spectrometry. Anal. Sci. 2011, 27, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Inoue, S.; Sugiura, Y.; Ueno, Y. Simultaneous determination of trichothecene mycotoxins and zaaralenone in cereal by gas chromatography mass spectrometry. J. Chromatogr. A 2000, 882, 23–28. [Google Scholar] [CrossRef]
- Ferreira, I.; Fernandes, J.O.; Cunha, S.C. Optimization and validation of a method based in a QuEChERS procedure and gas chromatography–mass spectrometry for the determination of multi-mycotoxins in popcorn. Food Control 2012, 27, 188–193. [Google Scholar] [CrossRef]
- Ndube, N.; van der Westhuizen, L.; Green, I.R.; Shephard, G.S. HPLC determination of fumonisin mycotoxins in maize: A comparative study of naphthalene-2,3-dicarboxaldehyde and o-phthaldialdehyde derivatization reagents for fluorescence and diode array detection. J. Chromatogr. B 2011, 879, 2239–2243. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.; MacDonald, S.J.; Boughtflower, V.; Brereton, P. Simultaneous determination of aflatoxins and ochratoxin A in food using a fully automated immunoaffinity column clean-up and liquid chromatography–fluorescence detection. J. Chromatogr. A 2004, 1059, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Romero-Gonzalez, R.; Garrido Frenich, A.; Martinez Vidal, J.L.; Prestes, O.D.; Grio, S.L. Simultaneous determination of pesticides, biopesticides and mycotoxins in organic products applying a quick, easy, cheap, effective, rugged and safe extraction procedure and ultra-high performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2011, 1218, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Zachariasova, M.; Lacina, O.; Malachova, A.; Kostelanska, M.; Poustka, J.; Godula, M.; Hajslova, J. Novel approaches in analysis of Fusarium mycotoxins in cereals employing ultra performance liquid chromatography coupled with high resolution mass spectrometry. Anal. Chim. Acta 2010, 662, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Arroyo-Manzanares, N.; Huertas-Pérez, J.F.; García-Campaña, A.M.; Gámiz-Gracia, L. Simple methodology for the determination of mycotoxins in pseudocereals, spelt and rice. Food Control 2014, 36, 94–101. [Google Scholar] [CrossRef]
- Malachova, A.; Sulyok, M.; Beltran, E.; Berthiller, F.; Krska, R. Optimization and validation of a quantitative liquid chromatography-tandem mass spectrometric method covering 295 bacterial and fungal metabolites including all regulated mycotoxins in four model food matrices. J. Chromatogr. A 2014, 1362, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Škrbić, B.; Đurišić-Mladenović, M.G.N.; Živančev, A.J. Multi-mycotoxin Analysis by UHPLC-HESI-MSMS: A Preliminary Survey of Serbian Wheat Flour A. Agron. Res. 2011, 9, 461–468. [Google Scholar]
- Vaclavik, L.; Zachariasova, M.; Hrbek, V.; Hajslova, J. Analysis of multiple mycotoxins in cereals under ambient conditions using direct analysis in real time (DART) ionization coupled to high resolution mass spectrometry. Talanta 2010, 82, 1950–1957. [Google Scholar] [CrossRef] [PubMed]
- Rubert, J.; Dzuman, Z.; Vaclavikova, M.; Zachariasova, M.; Soler, C.; Hajslova, J. Analysis of mycotoxins in barley using ultra high liquid chromatography high resolution mass spectrometry: Comparison of efficiency and efficacy of different extraction procedures. Talanta 2012, 99, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Desmarchelier, A.; Tessiot, S.; Bessaire, T.; Racault, L.; Fiorese, E.; Urbani, A.; Chan, W.C.; Cheng, P.; Mottier, P. Combining the quick, easy, cheap, effective, rugged and safe approach and clean-up by immunoaffinity column for the analysis of 15 mycotoxins by isotope dilution liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2014, 1337, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Li, W.; Zhang, Y.; Sun, L.; Dong, X.; Hu, X.; Wang, B. Simultaneous determination of twelve mycotoxins in cereals by ultra-high performance liquid chromatography-tandem mass spectrometry. Acta Agron. Sin. 2014, 40, 691–701. [Google Scholar] [CrossRef]
- Kirinčič, S.; Škrjanc, B.; Kos, N.; Kozolc, B.; Pirnat, N.; Tavčar-Kalcher, G. Mycotoxins in cereals and cereal products in Slovenia Official control of foods in the years 2008–2012. Food Control 2015, 50, 157–165. [Google Scholar] [CrossRef]
- Myresiotis, C.K.; Testempasis, S.; Vryzas, Z.; Karaoglanidis, G.S.; Papadopoulou-Mourkidou, E. Determination of mycotoxins in pomegranate fruits and juices using a QuEChERS-based method. Food Chem. 2015, 182, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Arroyo-Manzanares, N.; Huertas-Perez, J.F.; Gamiz-Gracia, L.; Garcia-Campana, A.M. A new approach in sample treatment combined with UHPLC-MS/MS for the determination of multiclass mycotoxins in edible nuts and seeds. Talanta 2013, 115, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, P.J.; Barros, N.; Câmara, J.S. A survey of the occurrence of ochratoxin A in Madeira wines based on a modified QuEChERS extraction procedure combined with liquid chromatography–triple quadrupole tandem mass spectrometry. Food Res. Int. 2013, 54, 293–301. [Google Scholar] [CrossRef]
- Rubert, J.; Leon, N.; Saez, C.; Martins, C.P.; Godula, M.; Yusa, V.; Manes, J.; Soriano, J.M.; Soler, C. Evaluation of mycotoxins and their metabolites in human breast milk using liquid chromatography coupled to high resolution mass spectrometry. Anal. Chim. Acta 2014, 820, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Han, S.; Lu, M.; Wang, P.; Han, J.; Wang, J. Modified QuEChERS method combined with ultra-high performance liquid chromatography tandem mass spectrometry for the simultaneous determination of 26 mycotoxins in sesame butter. J. Chromatogr. B 2014, 970, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Sirhan, A.Y.; Tan, G.H.; Wong, R.C.S. Method validation in the determination of aflatoxins in noodle samples using the QuEChERS method (Quick, Easy, Cheap, Effective, Rugged and Safe) and high performance liquid chromatography coupled to a fluorescence detector (HPLC–FLD). Food Control 2011, 22, 1807–1813. [Google Scholar] [CrossRef]
- Frenich, A.G.; Romero-Gonzalez, R.; Gomez-Perez, M.L.; Vidal, J.L. Multi-mycotoxin analysis in eggs using a QuEChERS-based extraction procedure and ultra-high-pressure liquid chromatography coupled to triple quadrupole mass spectrometry. J. Chromatogr. A 2011, 1218, 4349–4356. [Google Scholar] [CrossRef] [PubMed]
- Belen Serrano, A.; Capriotti, A.L.; Cavaliere, C.; Piovesana, S.; Samperi, R.; Ventura, S.; Lagana, A. Development of a Rapid LC-MS/MS Method for the Determination of Emerging Fusarium mycotoxins Enniatins and Beauvericin in Human Biological Fluids. Toxins 2015, 7, 3554–3571. [Google Scholar] [CrossRef] [PubMed]
- Rubert, J.; Soler, C.; Mañes, J. Application of an HPLC–MS/MS method for mycotoxin analysis in commercial baby foods. Food Chem. 2012, 133, 176–183. [Google Scholar] [CrossRef]
- Capriotti, A.L.; Cavaliere, C.; Foglia, P.; Samperi, R.; Stampachiacchiere, S.; Ventura, S.; Lagana, A. Multiclass analysis of mycotoxins in biscuits by high performance liquid chromatography-tandem mass spectrometry. Comparison of different extraction procedures. J. Chromatogr. A 2014, 1343, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Azaiez, I.; Giusti, F.; Sagratini, G.; Mañes, J.; Fernández-Franzón, M. Multi-mycotoxins Analysis in Dried Fruit by LC/MS/MS and a Modified QuEChERS Procedure. Food Anal. Methods 2014, 7, 935–945. [Google Scholar] [CrossRef]
- Koesukwiwat, U.; Sanguankaew, K.; Leepipatpiboon, N. Evaluation of modified QuEChERS method for analysis of mycotoxins in rice. Food Chem. 2014, 153, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Dzuman, Z.; Zachariasova, M.; Veprikova, Z.; Godula, M.; Hajslova, J. Multi-analyte high performance liquid chromatography coupled to high resolution tandem mass spectrometry method for control of pesticide residues, mycotoxins, and pyrrolizidine alkaloids. Anal. Chim. Acta 2015, 863, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Bryla, M.; Waskiewicz, A.; Podolska, G.; Szymczyk, K.; Jedrzejczak, R.; Damaziak, K.; Sulek, A. Occurrence of 26 Mycotoxins in the Grain of Cereals Cultivated in Poland. Toxins 2016, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Arroyo-Manzanares, N.; Huertas-Perez, J.F.; Gamiz-Gracia, L.; Garcia-Campana, A.M. Simple and efficient methodology to determine mycotoxins in cereal syrups. Food Chem. 2015, 177, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Moltó, J.-C.; El Akhdari, S.; Mañes, J.; Zinedine, A. Simultaneous determination of Fusarium mycotoxins in wheat grain from Morocco by liquid chromatography coupled to triple quadrupole mass spectrometry. Food Control 2014, 46, 1–5. [Google Scholar] [CrossRef]
- Ren, Y.; Zhang, Y.; Shao, S.; Cai, Z.; Feng, L.; Pan, H.; Wang, Z. Simultaneous determination of multi-component mycotoxin contaminants in foods and feeds by ultra-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2007, 1143, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Su, X.; Chen, F.; Wang, P.; Fan, X.; Zhang, W. Determination of 26 Mycotoxins in Feedstuffs by Multifunctional Cleanup Column and Liquid Chromatography-Tandem Mass Spectrometry. Chin. J. Anal. Chem. 2015, 43, 264–270. [Google Scholar] [CrossRef]
- Njumbe Ediage, E.; Van Poucke, C.; De Saeger, S. A multi-analyte LC-MS/MS method for the analysis of 23 mycotoxins in different sorghum varieties: The forgotten sample matrix. Food Chem. 2015, 177, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhang, Y.; Lai, S.; Han, Z.; Wu, Y. Simultaneous determination of fumonisins B1, B2 and B3 contaminants in maize by ultra high-performance liquid chromatography tandem mass spectrometry. Anal. Chim. Acta 2011, 692, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Bolechová, M.; Benešová, K.; Běláková, S.; Čáslavský, J.; Pospíchalová, M.; Mikulíková, R. Determination of seventeen mycotoxins in barley and malt in the Czech Republic. Food Control 2015, 47, 108–113. [Google Scholar] [CrossRef]
- Yogendrarajah, P.; Van Poucke, C.; De Meulenaer, B.; De Saeger, S. Development and validation of a QuEChERS based liquid chromatography tandem mass spectrometry method for the determination of multiple mycotoxins in spices. J. Chromatogr. A 2013, 1297, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oueslati, S.; Meca, G.; Mliki, A.; Ghorbel, A.; Mañes, J. Determination of Fusarium mycotoxins enniatins, beauvericin and fusaproliferin in cereals and derived products from Tunisia. Food Control 2011, 22, 1373–1377. [Google Scholar] [CrossRef]
- The Commission of the European Communities. Commission Regulation (EC) No 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs. Off. J. Eur. Unione 2006, L364, 5–24. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mycotoxin | 100 mg C18 | 100 mg C18 + 10 mg PSA | 100 mg C18 + 20 mg PSA | 100 mg C18 + 50 mg PSA | 100 mg C18 + 100 mg PSA | 150 mg PSA | 150 mg GCB | MycoSep 226 | MycoSep 227 | MycoSep 400 | SPE C18 | SPE NH2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DON | 82.6 | 96.5 | 76.8 | 85.1 | 89.0 | 61.3 | 58.8 | 110.0 | 86.9 | 87.5 | 88.2 | 94.1 |
| 15-DON | 79.2 | 81.3 | 77.1 | 84.0 | 78.4 | 34.9 | 18.7 | 125.1 | 113.8 | 74.8 | 78.7 | 90.3 |
| 3-DON | 85.3 | 88.4 | 68.7 | 86.9 | 85.4 | 30.3 | 15.8 | 106.3 | 100.7 | 77.4 | 112.1 | 112.1 |
| AF B1 | 67.7 | 67.9 | 44.7 | 17.1 | 24.2 | ND | ND | 73.5 | ND | 68.8 | 64.6 | 81.6 |
| AF B2 | 65.3 | 62.3 | 61.0 | 13.0 | 15.0 | ND | ND | 68.7 | ND | 63.2 | 71.9 | 45.0 |
| AF G1 | 64.7 | 61.2 | 64.5 | 32.7 | 14.7 | 3.2 | ND | 86.1 | ND | 63.9 | 67.3 | 36.4 |
| AF G2 | 101.7 | 72.0 | 103.3 | ND | ND | ND | ND | 71.6 | ND | 60.4 | 61.3 | 72.1 |
| ZEN | 67.0 | 50.0 | 18.9 | 43.8 | 23.5 | 22.6 | 0.7 | 74.8 | 0.9 | 53.9 | 30.7 | 49.4 |
| SMC | 52.1 | 38.4 | 12.7 | 40.8 | 39.6 | 19.6 | ND | 62.7 | ND | 36.6 | 34.0 | 51.4 |
| OTA | 71.8 | 8.7 | 23.2 | 2.6 | 0.7 | 21.5 | 0.5 | 15.4 | 4.1 | 40.1 | 71.9 | 5.1 |
| HT-2 | 36.3 | 50.1 | 8.8 | 42.1 | 41.5 | 13.6 | 8.6 | 70.6 | 72.9 | 67.2 | 29.2 | 33.2 |
| T-2 | 51.9 | 49.6 | 14.6 | 54.0 | 53.1 | 22.0 | 11.2 | 86.1 | 94.1 | 61.7 | 34.6 | 37.1 |
| VCG | 63.6 | 67.6 | 60.4 | 68.7 | 85.2 | 28.1 | 1.0 | 69.7 | 65.2 | 61.3 | 26.3 | 40.2 |
| ENA | 82.2 | 39.1 | 16.4 | 34.1 | 29.3 | 16.8 | 4.1 | 6.1 | ND | 25.2 | 6.4 | 65.0 |
| ENA1 | 60.0 | 30.8 | 15.9 | 23.3 | 24.3 | 26.8 | 8.1 | 8.2 | ND | 28.6 | 14.4 | 66.0 |
| ENB | 64.4 | 49.5 | 47.1 | 38.0 | 41.8 | 35.2 | 15.2 | 30.6 | ND | 39.1 | 32.1 | 83.6 |
| ENB1 | 60.4 | 33.9 | 24.8 | 28.6 | 30.7 | 35.4 | 12.1 | 17.6 | ND | 36.1 | 22.2 | 74.5 |
| BEA | 66.2 | 33.2 | 34.6 | 30.4 | 47.5 | 16.2 | 3.4 | 35.5 | ND | 27.2 | 13.9 | 61.3 |
| FUS-X | 62.2 | 79.4 | 83.3 | 67.6 | 71.3 | 19.2 | 10.3 | 100.2 | 94.8 | 71.1 | 91.3 | 77.0 |
| GLT | 98.6 | 82.3 | 56.3 | 75.2 | 66.6 | 26.7 | 11.5 | 21.5 | 5.0 | 24.7 | 69.1 | 5.5 |
| NEO | 28.9 | 26.0 | 17.4 | 27.2 | 25.2 | 5.3 | 13.7 | 58.4 | 37.3 | 42.8 | 14.0 | 31.0 |
| DAS | 45.9 | 38.0 | 49.1 | 21.8 | 34.9 | 31.6 | 9.8 | 115.5 | 108.1 | 85.1 | 65.9 | 51.1 |
| FB1 | 63.7 | 3.9 | 2.5 | 1.3 | 0.5 | 0.2 | 7.6 | 2.4 | 0.8 | 1.5 | 117.0 | 3.5 |
| FB2 | 89.5 | 8.2 | 5.1 | 3.0 | 1.4 | 0.6 | 4.9 | 6.4 | 0.6 | 4.2 | 113.3 | 1.1 |
| FB3 | 84.5 | 5.6 | 4.3 | 3.0 | 1.8 | 0.9 | 8.5 | 2.8 | 0.9 | 11.7 | 114.4 | 3.6 |
| Compound | Calibration Curve | Linear Range | r | LOD | LOQ | Intra-Day Precision (n = 6) | Inter-Day Precision (n = 6) |
|---|---|---|---|---|---|---|---|
| (µg·kg−1) | (µg·kg−1) | (µg·kg−1) | RSD (%) | RSD (%) | |||
| DON | Y = 98.62X + 169.00 | 5.0~500 | 0.9995 | 5.0 | 15.0 | 3.4 | 7.0 |
| 15-DON | Y = 313.95X + 1394.48 | 5.0~500 | 0.9997 | 2.0 | 5.0 | 3.6 | 7.1 |
| 3-DON | Y = 191.02X + 375.01 | 5.0~500 | 0.9999 | 2.0 | 5.0 | 3.3 | 7.3 |
| AF B1 | Y = 853.62X + 1079.28 | 0.1~50 | 0.9950 | 0.03 | 0.1 | 8.9 | 7.2 |
| AF B2 | Y = 1284.87X + 470.48 | 0.3~15 | 0.9974 | 0.1 | 0.3 | 5.3 | 9.2 |
| AF G1 | Y = 945.23X + 253.68 | 0.1~50 | 0.9999 | 0.1 | 0.3 | 8.1 | 12.2 |
| AF G2 | Y = 378.07X ± 111.02 | 0.3~15 | 0.9960 | 0.1 | 0.3 | 8.8 | 16.9 |
| ZEN | Y = 272.06X ± 695.88 | 1.0~200 | 0.9993 | 1.0 | 4.0 | 6.4 | 6.5 |
| SMC | Y = 2071.13X + 1706.52 | 0.4~200 | 0.9998 | 0.1 | 0.4 | 4.2 | 9.6 |
| OTA | Y = 573.77X ± 2398.65 | 0.4~200 | 0.9997 | 0.1 | 0.4 | 8.4 | 9.3 |
| HT-2 | Y = 174.01X ± 181.35 | 0.4~200 | 0.9996 | 0.1 | 0.4 | 8.3 | 7.9 |
| T-2 | Y = 1144.87X + 2500.6 | 0.4~200 | 0.9996 | 0.1 | 0.4 | 6.1 | 7.2 |
| VCG | Y = 581.24X + 1550.22 | 0.2~200 | 0.9997 | 0.1 | 0.4 | 8.2 | 7.4 |
| ENNA | Y = 222.75X ± 123.09 | 0.2~200 | 0.9998 | 0.05 | 0.2 | 7.8 | 12.6 |
| ENNA1 | Y = 6333.74X + 5213.22 | 0.2~200 | 0.9998 | 0.05 | 0.2 | 2.4 | 11.1 |
| ENNB | Y = 6124.97X ± 673.19 | 0.1~200 | 0.9998 | 0.05 | 0.2 | 5.6 | 12.9 |
| ENNB1 | Y = 4212.47X + 2487.88 | 0.1~200 | 0.9997 | 0.05 | 0.2 | 6.9 | 14.7 |
| BEA | Y = 3170.66X + 659.08 | 0.1~200 | 0.9999 | 0.05 | 0.2 | 7.3 | 9.3 |
| FUS-X | Y = 28.65X ± 15.62 | 0.5~500 | 0.9996 | 10.0 | 25.0 | 6.9 | 9.4 |
| GLT | Y = 473.91X + 2091.75 | 5.0~200 | 0.9993 | 5.0 | 10.0 | 4.6 | 6.1 |
| NEO | Y = 97.93X + 1000.67 | 5.0~200 | 0.9993 | 5.0 | 10.0 | 7.0 | 17.6 |
| DAS | Y = 97.93X + 1000.67 | 10.0~500 | 0.9993 | 10.0 | 25.0 | 5.6 | 14.3 |
| FB1 | Y = 83.71X ± 323.38 | 25.0~500 | 0.9992 | 15.0 | 25.0 | 6.3 | 6.3 |
| FB2 | Y = 70.66X ± 421.58 | 25.0~500 | 0.9950 | 15.0 | 25.0 | 12.0 | 8.5 |
| FB3 | Y = 60.71X + 2650.23 | 25.0~500 | 0.9995 | 15.0 | 25.0 | 7.4 | 7.8 |
| Mycotoxin | Wheat (n = 6) | Corn (n = 6) | Rice (n = 6) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Level 1 | Level 2 | Level 3 | Level 1 | Level 2 | Level 3 | Level 1 | Level 2 | Level 3 | |
| DON | 115.8 (0.7) | 97.9 (5.5) | 98.8 (6.8) | 98.4 (15.8) | 106.6 (5.9) | 101.2 (1.9) | 97.0 (4.7) | 116.9 (5.3) | 106.5 (1.0) |
| 15-ADON | 91.0 (3.6) | 100.8 (4.0) | 96.8 (2.6) | 88.9 (3.4) | 95 (2.5) | 97.9 (0.5) | 85.8 (3.9) | 113.1 (3.4) | 93.0 (2.5) |
| 3-ADON | 93.4 (2.9) | 104.7 (1.4) | 96.8 (1.7) | 85 (9.7) | 93.8 (4.3) | 102.5 (4.5) | 73.4 (7.2) | 100.6 (0.5) | 80.9 (2.8) |
| AF B1 | 61.7 (10.8) | 118.1 (1.7) | 87.4 (12.2) | 82.3 (9.3) | 60.3 (18.5) | 90 (5.4) | 82.8 (7.0) | 91.7 (17.1) | 105.0 (5.1) |
| AF B2 | 114.0 (18.1) | 63.0 (6.2) | 71.0 (12.0) | 60.6 (11.1) | 108.3 (14.9) | 91.6 (12.9) | 67.9 (6.0) | 98.3 (4.7) | 79.4 (19.0) |
| AF G1 | 84.8 (10.5) | 73.2 (12.8) | 88.2 (14.8) | 73.4 (18.8) | 80.7 (8.5) | 86.7 (3.4) | 88.8 (8.1) | 89.4 (19.2) | 95.9 (18.6) |
| AF G2 | 75.7 (11.4) | 91.5 (9.5) | 84.5 (5.8) | 114.7 (13.4) | 89.9 (17.1) | 63.7 (19.6) | 62.5 (11.8) | 103.3 (16.5) | 100.4 (8.7) |
| ZEN | 70.0 (6.4) | 73.5 (8.5) | 63.3 (12.2) | 92 (4.4) | 68.1 (7.3) | 88.6 (5.3) | 84.6 (17.1) | 64.3 (10.5) | 79.2 (2.1) |
| SMC | 64.8 (10.0) | 62.8 (5.8) | 69.9 (8.4) | 69.1 (4.7) | 69.1 (3.6) | 63.5 (9.1) | 66.4 (10.5) | 58.2 (1.5) | 86.2 (6.2) |
| OTA | 77.4 (1.4) | 88.7 (11.2) | 60.1 (2.0) | 77.8 (7.0) | 65.7 (2.7) | 65.5 (7.7) | 74.0 (8.7) | 68.6 (5.9) | 81.4 (9.5) |
| HT-2 | 77.5 (5.1) | 73.0 (11.8) | 84.1 (8.3) | 73.6 (0.8) | 70.8 (10.3) | 89.7 (2.8) | 83.8 (9.2) | 103.6 (4.4) | 86.1 (3.3) |
| T-2 | 62.4 (8.8) | 67.9 (2.5) | 69.9 (2.0) | 77.0 (5.7) | 69.4 (4.0) | 85.9 (4.3) | 82.2 (3.6) | 122.4 (0.6) | 85.0 (1.5) |
| VCG | 61.4 (5.5) | 106.5 (6.7) | 62.4 (6.1) | 67.6 (8.4) | 71.9 (10.1) | 113.2 (0.2) | 92.7 (1.4) | 79.0 (7.1) | 89.5 (9.0) |
| ENNA | 71.2 (9.1) | 63.5 (1.5) | 67.0 (9.2) | 65.7 (6.9) | 63.5 (4.0) | 89.2 (0.6) | 73.9 (5.2) | 69.3 (10.6) | 99.0 (1.1) |
| ENNA1 | 74.5 (3.5) | 64.9 (5.3) | 61.9 (8.9) | 67.2 (4.4) | 63.7 (4.9) | 75.6 (0.7) | 82.3 (11.7) | 74.2 (4.4) | 73.4 (6.7) |
| ENNB | 67.4 (14.3) | 69.7 (5.3) | 70.2 (5.1) | 71.4 (8.3) | 96.6 (13.0) | 81.7 (8.7) | 104.2 (6.7) | 79.3 (2.7) | 87.7 (3.9) |
| ENNB1 | 67.7 (12.2) | 65.0 (11.2) | 60.2 (5.0) | 64.9 (9.2) | 73.1 (1.1) | 76.2 (15.4) | 80.0 (11.4) | 77.3 (2.8) | 79.1 (1.0) |
| BEA | 65.4 (7.6) | 71.7 (3.3) | 60.3 (3.2) | 66.5 (3.5) | 74.8 (3.9) | 65.1 (19.6) | 72.8 (9.4) | 67.9 (1.6) | 62.5 (4.1) |
| FUS-X | 97.5 (14.5) | 107.9 (4.9) | 119.3 (11.7) | 101.4 (4.5) | 87.2 (10.) | 111.2 (10.6) | 76.5 (3.6) | 110.4 (19.0) | 92.1 (6.9) |
| GLT | 67.7 (1.3) | 74.3 (5.2) | 82.4 (2.6) | 75.7 (3.2) | 75.7 (1.0) | 79.3 (4.4) | 69.9 (3.9) | 86.3 (2.5) | 74.9 (1.2) |
| NEO | 63.7 (9.5) | 69.3 (12.5) | 64.6 (1.1) | 63.2 (18.5) | 95.3 (15.8) | 62.6 (15.9) | 64.6 (7.6) | 108.7 (14.2) | 67.2 (10.9) |
| DAS | 77.0 (12.2) | 84.0 (14.3) | 100.7 (4.1) | 70.8 (2.9) | 88.1 (1.4) | 87.8 (11.7) | 78.8 (17.4) | 72.6 (4.1) | 88.4 (2.5) |
| FB1 | 109.4 (4.5) | 107.1 (7.8) | 73.9 (2.3) | 118.9 (2.1) | 115.4 (2.8) | 83.7 (4.7) | 107.8 (3.6) | 113.7 (2.8) | 80.2 (1.5) |
| FB2 | 102.3 (3.2) | 102.4 (6.9) | 74.7 (2.4) | 107.4 (4.9) | 110.8 (5.6) | 87.8 (2.1) | 100.9 (0.7) | 99.5 (2.4) | 105.7 (2.0) |
| FB3 | 115.6 (5.2) | 102.8 (5.9) | 89.6 (1.9) | 115.5 (2.3) | 114 (2.7) | 89.1 (3.5) | 111.2 (5.4) | 107.4 (3.7) | 91.3 (1.1) |
| Mycotoxins | Retention Time (min) | Precursor Ion (m/z) | Product Ion (m/z) | Dwell Time (s) | Cone Voltage (V) | Collision Energy (V) |
|---|---|---|---|---|---|---|
| DON | 3.22 | 297.28 | 249.1 (Q) | 0.005 | 20 | 10 |
| 203.1 (q) | 0.005 | 20 | 14 | |||
| 15-ADON | 4.38 | 339.3 | 137.1 (Q) | 0.005 | 22 | 10 |
| 231.2 (q) | 0.005 | 22 | 12 | |||
| 3-ADON | 4.37 | 339.2 | 231.2 (Q) | 0.005 | 20 | 14 |
| 213.2 (q) | 0.005 | 20 | 22 | |||
| AF B1 | 5.13 | 313.28 | 241.1 (Q) | 0.005 | 46 | 38 |
| 285.0 (q) | 0.005 | 46 | 22 | |||
| AF B2 | 4.97 | 315.2 | 259.1 (Q) | 0.005 | 40 | 30 |
| 287.1 (q) | 0.005 | 40 | 26 | |||
| AF G1 | 4.78 | 329.03 | 199.8 (Q) | 0.005 | 40 | 40 |
| 243.0 (q) | 0.005 | 40 | 28 | |||
| AF G2 | 4.63 | 331.29 | 189.1 (Q) | 0.005 | 44 | 34 |
| 245.1 (q) | 0.005 | 44 | 28 | |||
| ZEN | 6.45 | 319.05 | 283.0 (Q) | 0.005 | 18 | 22 |
| 187.3 (q) | 0.005 | 18 | 14 | |||
| SMC | 6.63 | 325.20 | 281.0 (Q) | 0.005 | 45 | 36 |
| 253.1 (q) | 0.005 | 45 | 42 | |||
| OTA | 6.43 | 404.24 | 239.1 (Q) | 0.005 | 26 | 36 |
| 221.1 (q) | 0.005 | 26 | 26 | |||
| HT-2 | 5.75 | 447.30 | 345.2 (Q) | 0.005 | 34 | 18 |
| 285.1 (q) | 0.005 | 34 | 22 | |||
| T-2 | 6.08 | 489.31 | 245.1 (Q) | 0.005 | 42 | 34 |
| 387.2 (q) | 0.005 | 42 | 22 | |||
| VCG | 6.73 | 534.32 | 392.2 (Q) | 0.005 | 22 | 18 |
| 191.1 (q) | 0.005 | 22 | 24 | |||
| ENNA | 7.66 | 682.29 | 210.0 (Q) | 0.005 | 48 | 26 |
| 228.0 (q) | 0.005 | 48 | 28 | |||
| ENNA1 | 7.50 | 668.28 | 99.9 (Q) | 0.005 | 48 | 60 |
| 210.0 (q) | 0.005 | 48 | 26 | |||
| ENNB | 7.30 | 640.18 | 195.9 (Q) | 0.005 | 46 | 24 |
| 213.9 (q) | 0.005 | 46 | 26 | |||
| ENNB1 | 7.45 | 654.26 | 99.9 (Q) | 0.005 | 46 | 54 |
| 195.9 (q) | 0.005 | 46 | 24 | |||
| BEA | 7.36 | 784.24 | 243.9 (Q) | 0.005 | 48 | 28 |
| 262.0 (q) | 0.005 | 48 | 26 | |||
| FUS-X | 3.74 | 354.84 | 174.8 (Q) | 0.005 | 18 | 34 |
| 136.8 (q) | 0.005 | 18 | 34 | |||
| GLT | 5.22 | 326.81 | 262.9 (Q) | 0.005 | 16 | 10 |
| 244.9 (q) | 0.005 | 16 | 16 | |||
| NEO | 3.82 | 399.93 | 214.9 (Q) | 0.005 | 16 | 18 |
| 304.9 (q) | 0.005 | 16 | 14 | |||
| DAS | 5.18 | 383.94 | 306.9 (Q) | 0.005 | 18 | 12 |
| 246.9 (q) | 0.005 | 18 | 16 | |||
| FB1 | 5.56 | 722.35 | 334.2 (Q) | 0.005 | 56 | 42 |
| 352.2 (q) | 0.005 | 56 | 36 | |||
| FB2 | 6.10 | 706.36 | 318.3 (Q) | 0.005 | 56 | 40 |
| 336.3 (q) | 0.005 | 56 | 38 | |||
| FB3 | 5.87 | 706.48 | 336.3 (Q) | 0.005 | 44 | 32 |
| 95.0 (q) | 0.005 | 44 | 48 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, J.; Li, W.; Zhang, Y.; Hu, X.; Wu, L.; Wang, B. QuEChERS Purification Combined with Ultrahigh-Performance Liquid Chromatography Tandem Mass Spectrometry for Simultaneous Quantification of 25 Mycotoxins in Cereals. Toxins 2016, 8, 375. https://doi.org/10.3390/toxins8120375
Sun J, Li W, Zhang Y, Hu X, Wu L, Wang B. QuEChERS Purification Combined with Ultrahigh-Performance Liquid Chromatography Tandem Mass Spectrometry for Simultaneous Quantification of 25 Mycotoxins in Cereals. Toxins. 2016; 8(12):375. https://doi.org/10.3390/toxins8120375
Chicago/Turabian StyleSun, Juan, Weixi Li, Yan Zhang, Xuexu Hu, Li Wu, and Bujun Wang. 2016. "QuEChERS Purification Combined with Ultrahigh-Performance Liquid Chromatography Tandem Mass Spectrometry for Simultaneous Quantification of 25 Mycotoxins in Cereals" Toxins 8, no. 12: 375. https://doi.org/10.3390/toxins8120375
APA StyleSun, J., Li, W., Zhang, Y., Hu, X., Wu, L., & Wang, B. (2016). QuEChERS Purification Combined with Ultrahigh-Performance Liquid Chromatography Tandem Mass Spectrometry for Simultaneous Quantification of 25 Mycotoxins in Cereals. Toxins, 8(12), 375. https://doi.org/10.3390/toxins8120375