1. Introduction
Corn (
Zea mays L.) is the most intensively cultivated cereal crop in the world, mainly due to its adaptive nature. It can be cultivated in both tropical and temperate climates and from sea level up to
ca. 3.5 km. The economic importance of corn is also reflected in its diverse use. While most harvested corn is used to prepare animal feed, use of corn derivatives is quite important in food production. In Brazil, corn is an important food source for many people living in the semi-arid area of the Northeastern Region [
1].
Fungi are ubiquitous microorganisms known to produce a wide variety of secondary metabolites, which play important role in diversification and adaptation of these microorganisms to various ecological niches, including plants cultivated for food and feed production, such as the corn plant and kernels [
2,
3,
4,
5,
6,
7,
8]. Secondary metabolites have attracted huge interest of various researchers due to their potential for biotechnological applications such as development of drugs, cosmetics, food, and others [
9]. However, one of the main concerning related with some fungal secondary metabolites is their toxicity.
Fusarium verticillioides Sacc. Niremberg (= Fusarium moniliforme Sheldon) is a non-obligatory parasite that commonly occurs in corn plants. It can cause damage to the roots and stem and spoilage in the produced corn kernels. Infection by this fungus is often asymptomatic. This species produces a set of mycotoxins and is considered one of the most globally significant sources of contamination by fumonisins in food products, especially in corn kernels.
There are 28 known chemical compounds of natural occurrence produced by filamentous fungi that are analogues of fumonisins. These compounds can be divided into four groups, identified as the A, B, C and P series. From a toxicological point of view, the B series (FB
1, FB
2 and FB
3) is the most relevant one, and fumonisins belonging to this series are common natural contaminants in corn [
10].
Genetic studies suggest that three
loci in
F. verticillioides are closely related to fumonisin synthesis. In this case,
FUM1 gene would be responsible for controlling the productive capacity of fumonisins by the fungus, while
FUM2 and
FUM3 would be responsible for controlling the hydroxylation of the C-10 and C-05 in the fumonisin molecule. Although this taxon is the most prolific producer of fumonisins, some
F. verticillioides isolates do not present the
FUM1 gene. This means that
F. verticillioides strains missing the
FUM1 gene are not potential producers of fumonisins [
10].
The
FUM1 gene, and other genes of this biosynthetic pathway, has been detected in
F. verticillioides isolates through the use of PCR gene amplification techniques [
11,
12,
13,
14]. In order to detect the presence of fungi in plant materials, specific primers in PCR reactions have widely been used. This technique is rapid, relatively easy to perform, quite sensitive, and can be used at wide or limited scales to examine the genetic structures of fungal populations [
12,
15].
A number of molecular techniques have been employed to characterise different species of
Fusarium. It includes PCR based on species-specific primers and the inter-simple sequence repeat (ISSR), which are molecular markers to detect genetic variability among populations [
12,
15,
16,
17].
In addition, matrix-assisted laser desorption/ionisation time-of-flight (MALDI-TOF) is a mass spectrometry technique that has demonstrated high potentiality on the identification of filamentous fungi at species and, in some specific cases, at strain level [
18]. One of the most interesting advantages of the technique is the possibility of analysing the intact fungal cell generating specific molecular mass profiles [
5,
19,
20,
21,
22].
As previously described in different studies, MALDI-TOF MS analysis for fungal identification is based on specific molecular masses that are mainly constituted by ribosomal proteins [
5,
20,
22]. The mass peaks generated by the analysis of these ribosomal proteins are observed in a mass range between 2 and 20 kDa. Also, compounds such as polysaccharides, lipids, phospholipids, and chitin, among others, can similarly be found in the fungal cell. All of these are very useful in fungal identifications by mass spectrometry [
5,
18,
19,
21,
23,
24].
Studies have demonstrated the high potentiality of the technique for species and strain identification within the
Fusarium genus [
24,
25,
26]. Recently, Santos
et al. [
27] used MALDI-TOF MS to establish a multistep identification of
Fusarium guttiforme and its antagonist
Trichoderma asperellum infecting pineapple side shoots. In addition, Rodrigues
et al. [
18] evaluated the MALDI-TOF MS technique regarding its capability for the species identification of fungal strains belonging to the genus
Aspergillus section
Flavi and their characterisation as aflatoxigenic or non-aflatoxigenic strains. According to the authors, the method was useful for fungal species differentiation. However, it was not able to differentiate strains according their mycotoxigenic potential.
The presence of fungal species such as
F. verticillioides, which is capable of producing mycotoxins in agricultural commodities, is a constant threat to both human and animal health. Efficient techniques of control that must include studies of inter- and intra-specific variations of this taxon and their capacities to produce mycotoxins are required [
10].
MALDI TOF tandem mass spectrometry (MS/MS) has been used as an important analytical tool in the analysis of low molecular weight physiological or therapeutic molecules of interest, such as amino acids and other pharmaceuticals [
28]. However, the application of MALDI-TOF MS/MS for the detection of fumonisins has not been addressed yet in the scientific literature.
As corn kernels are one of the main food and feed resources in the Brazilian Northeastern Region, and based on the lack of studies and information focusing the occurrence of mycotoxigenic F. verticillioides strains in this food and feed commodity produced in the Brazilian State of Pernambuco, the aim of the present study was to investigate the presence of mycotoxigenic F. verticillioides strains isolated from corn kernels in 3 different Regions of this State. For this propose, a polyphasic approach including classical taxonomy, molecular biology, MALDI-TOF MS and MALDI-TOF MS/MS for the identification and characterisation of the F. verticillioides strains was used.
3. Discussion
The results obtained herein demonstrated the appropriateness of the species-specific primers VERT-1 and VERT-2 used for confirming the identification of F. verticillioides isolates previously identified by classical morphology. All of the evaluated isolates presented 800 bp amplification products, as expected for this species. In fact, phenotypic methods are still relevant in daily routing analysis in a laboratory of mycology, but these methods are frequently associated with misidentification of some taxa. This is specially certainty for species belonging to the Fusarium genus, as their micromorphology can vary according to the culture medium employed.
Due to morphological variations within the genus
Fusarium and due to the fact that some species of the genera
Cylindrocarpon and
Acremonium are also able to produce fusiform and multicellular conidia, similar to those produced by
Fusarium species, molecular methods can significantly support confirmation of the taxonomy of that genus [
27,
30].
The data obtained in the present study are in agreement with the data presented by Dissanayake
et al. [
11], who evaluated fungal strains isolated from seeds and plants of Welsh Onion (
Allium fistulosum) in Japan. Authors successfully used the species-specific primers VERT-1 and VERT-2 to differentiate strains of
F.
verticillioides and further used the primers PRO1 (5′-CTTTCCGCCAAGTTTCTTC-3′) and PRO2 (5′-TGTCAGTAACTCGACGTTGTTG-3′) to differentiate strains of
F.
proliferatum.
A number of molecular biology techniques have been used to characterise different species of
Fusarium, including species-specific PCR primers. In order to prevent the early food contamination by fumonisins, González-Jáen
et al. [
17] evaluated the species-specific primers VERT-1 and VERT-2 as fast and reliable system for the detection of
F.
verticillioides able to produce these fungal secondary metabolites.
In addition, in order to identify
Fusarium species from corn kernels in India, Sreenivasa
et al. [
15] used the primer VERT-R (5′-CGACTCACGGCCAGGAAACC- 3′) combined with the primer VERTF-1 (5′-GCGGGAATTCAAAAGTGGCC-3′). The authors reported that 83 strains out of the 103 evaluated were identified at the species level as
F. verticillioides.
Some
F. verticillioides strains have been described as non-fumonisin producers [
12,
14]. Sánchez-Rangel
et al. [
13] evaluated 34
F. verticillioides strains isolated from corn in Mexico and found a correlation between the fumonisins production and the presence of the
FUM1 gene. The authors conclude that almost all of the
F. verticillioides strains that were fumonisin producers presented the
FUM1 gene.
In fact, the
FUM1 gene has widely been used as a molecular marker for the determination of mycotoxigenic
F. verticillioides strains. Maheshwar
et al. [
12] used successfully the primers VERT-1 and VERT-2 to distinguish fumonisin producers from non-producers strains of
F. verticillioides in 90 samples of stored paddy (
Oryza sativa L.), collected from different geographical regions of Karnataka, India.
In addition, Silva
et al. [
14] analysed 27
F. verticillioides strains isolated from corn and sorghum in Brazil. According to the authors, among the evaluated strains, 6 presented the
FUM1 gene. Karthikeyan
et al. [
31] evaluated 82 strains belonging to different
Fusarium species isolated from rice samples contaminated with fumonisins. In order to distinguish fumonisin producers from non-producers strains authors assessed fungal isolates by using PCR analyses through the primers VERTF-1 and VERTF-2. Twenty-one out 46
F. verticillioides strains presented amplification for the
FUM1 gene.
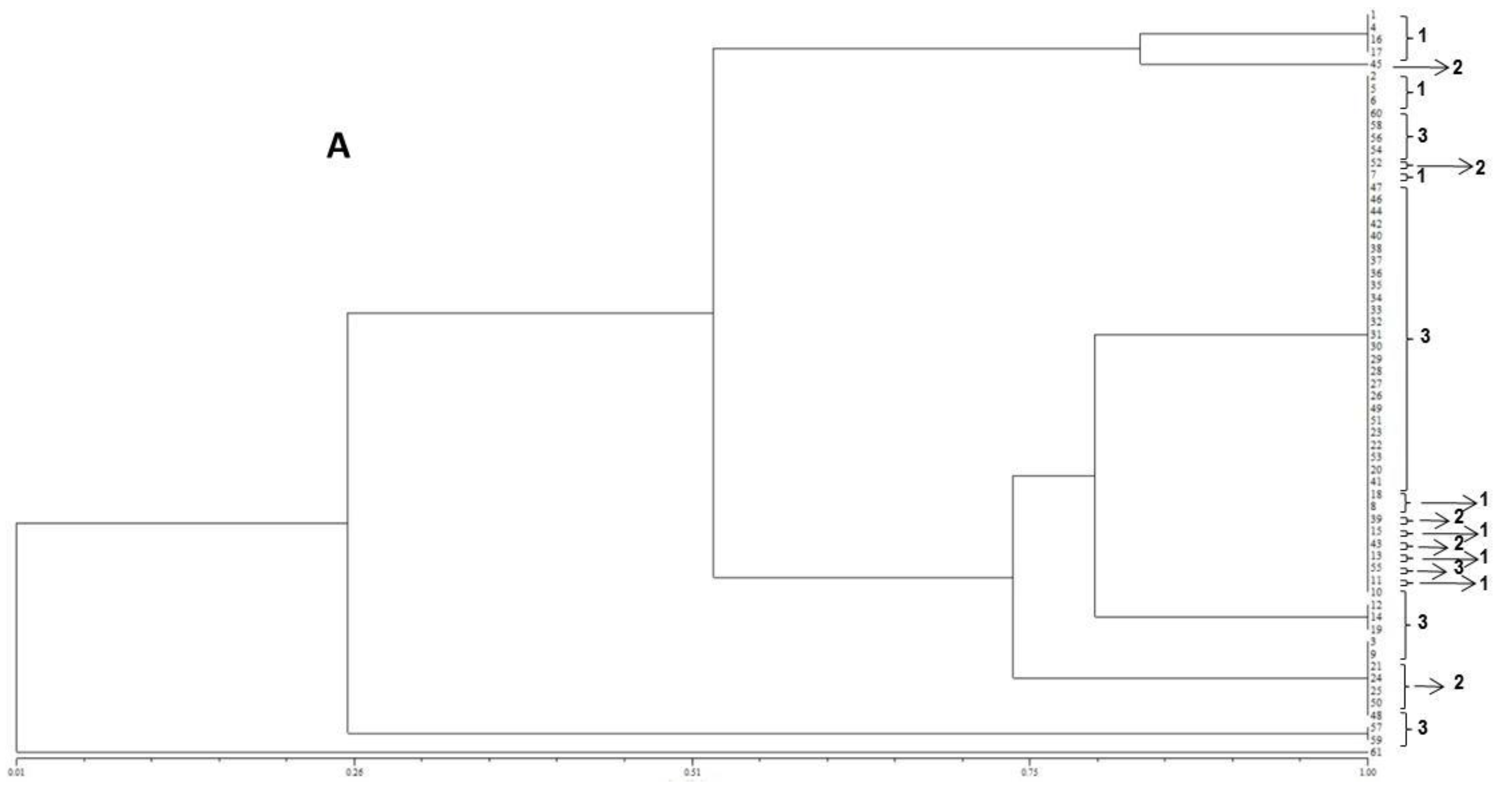
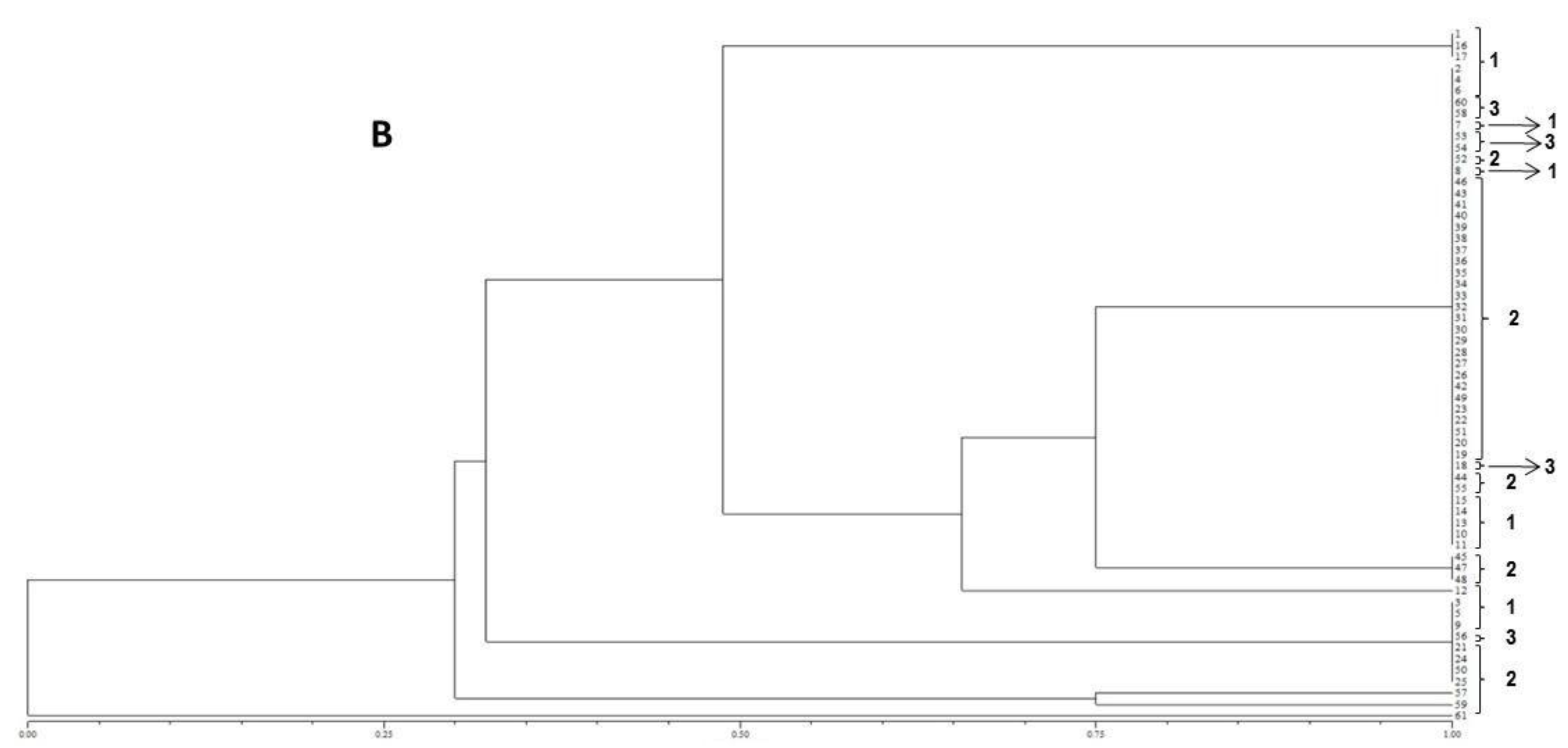
In the present study, the FUM1 gene was detected for all of the F. verticillioides strains assessed, confirming their potentials to produce fumonisins. The statistical analysis of the gels assembled using the primers (GTG)5 and (GACA)4 point out to a high genetic homogeneity among the different isolates. In terms of the geographical origins no groupings were observed for the assessed isolates. However, high levels of similarity among isolates were observed when ISSR primers were used. Furthermore, the (GTG)5 primer showed the highest genetic variability among them.
Few works have reported the use of ISSR primers for evaluation of
F. verticillioides strains. Lima [
32] reported that the (GTG)
5 primer was efficient for detecting differences among
Metarhizium anisopliae strains. Based on data obtained by the (GACA)
4 primer, the author found significant genetic variability among mutants and wild types strains belonging to the genus
Metarhizium. According to the author, the genetic variability found among the assessed strains allows their adaptation to the different hosts and wide geographic regions. In addition, Bayraktar
et al. [
16] evaluated 74 strains of
F. oxysporum f. sp.
ciceris using 20 ISSR primers. The authors reported that the genetic variability of the evaluated strains was greater according to the geographical regions of each evaluated strain.
Several studies have been proven the capability of MALDI-TOF MS to identify fungal species belonging to different fungal genus. MALDI-TOF MS has been proven to be useful for the identification of filamentous fungi at either the species or strain level [
18,
19,
33]. Moreover, previous studies have demonstrated the usefulness of MALDI-TOF MS-based fingerprinting methodology for the identification of species of
Fusarium [
24,
25,
26,
27].
By comparison with molecular biology data, MALDI-TOF MS has herein been shown to be sensitive (100% correct identification) for
F. verticillioides identification at the species level. The MALDI-TOF MS technique can thus be regarded as an additional step to the polyphasic scheme of identification of this fungal species. Furthermore, it is an objective and fast analytical methodology, and inexpensive in terms of labour and consumables when compared to other molecular biological techniques. As a consequence, it is suitable for applications which have particular high-throughput needs [
18]. According to the results obtained in the present study, no differences according to the geographical origin of the fungal strains were observed, which is in accordance with the molecular biology data obtained.
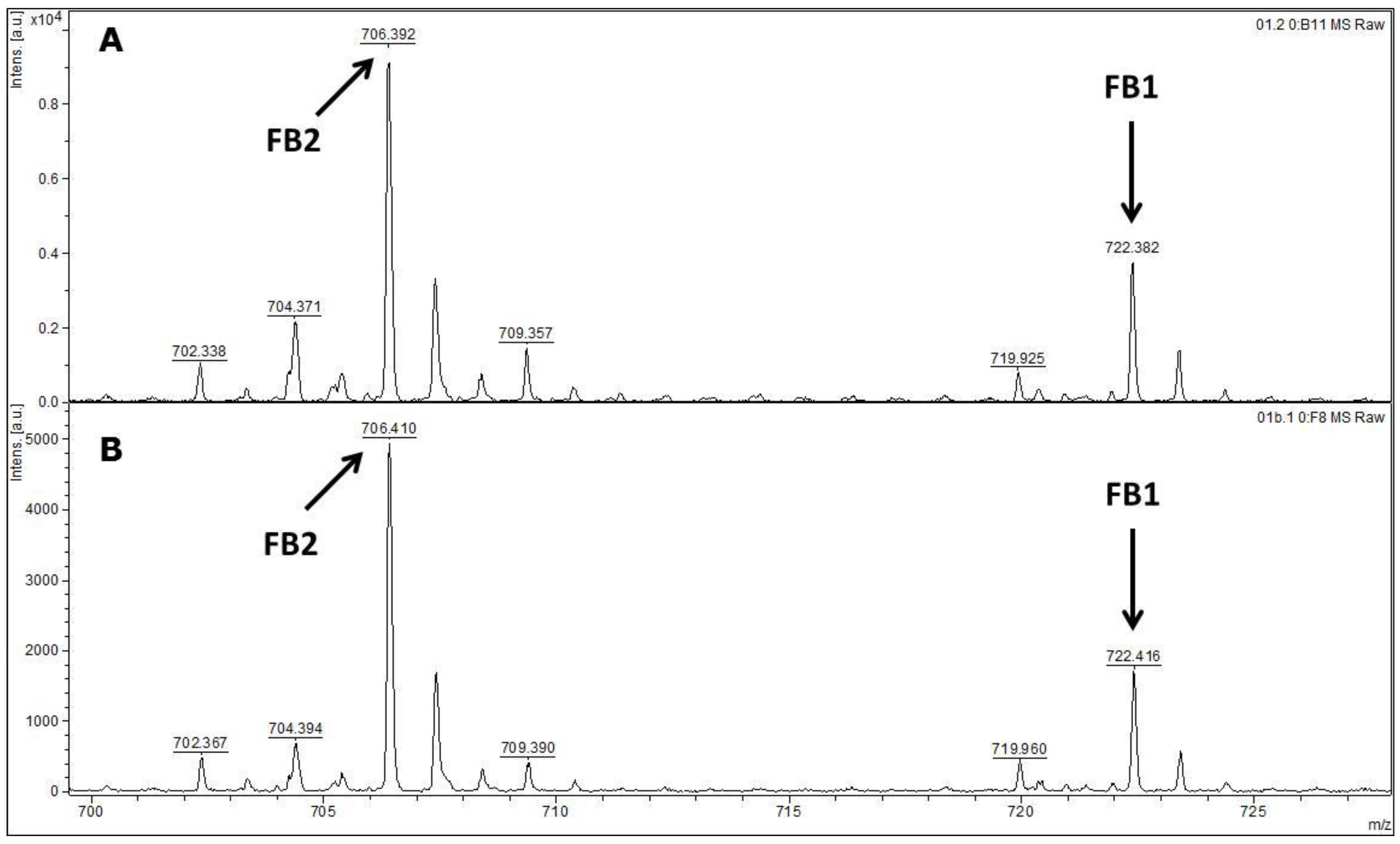
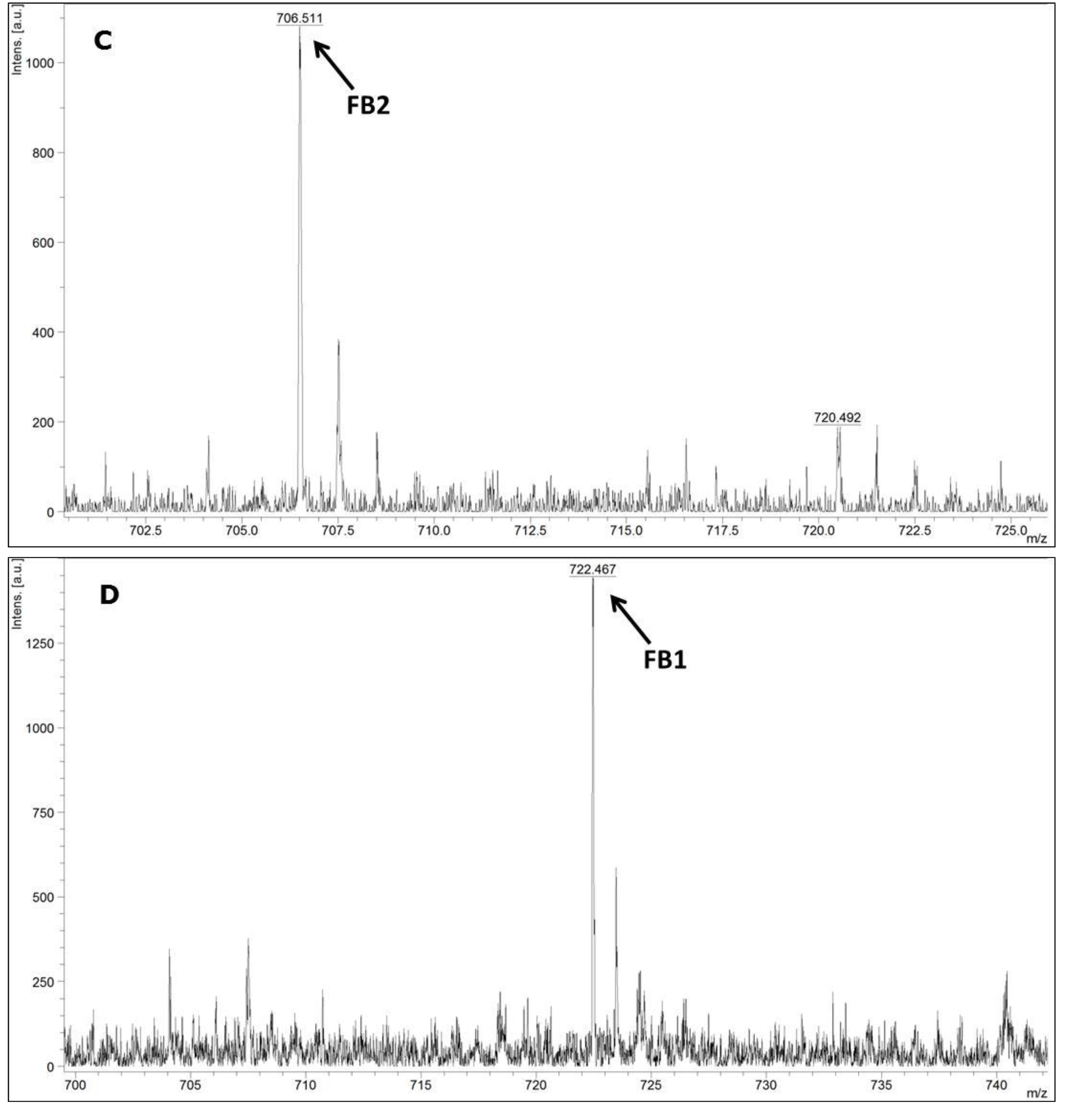
MALDI-TOF MS/MS was used in the present study to evaluate the production of fumonisin B1 and B2. These mycotoxins have been usually evaluated by other analytical techniques, such as HPLC or liquid chromatography coupled with mass spectrometry (e.g., LC-MS/MS) [
34,
35]. According to the data available in the scientific literature, there is no information about the use of MALDI-TOF MS/MS for the detection of fumonisin B1 and B2 in moistened ground corn samples obtained from corn kernels, which make this study the first report on the use of this analytical technique for this purpose. The results obtained herein for mycotoxin detection by MALDI-TOF MS/MS are in agreement with the data obtained by molecular biology based on the
FUM1 gene amplification, where all of the 60 strains evaluated were characterised as potential fumonisin B1 and B2 producers.
In conclusion, the results obtained in the present work point to a low genetic variability among the F. verticillioides strains isolated from corn kernels in different places in the 3 assessed Regions of the Brazilian State of Pernambuco. Furthermore, it was not possible to distinguish the F. verticillioides strains based on their origins, which suggest a homogeneous and very well adapted population. The MALDI-TOF technique was as good as molecular biology for the identification of F. verticillioides. In addition, MALDI-TOF MS/MS was reliable for the detection of fumonisin B1 and B2. Moreover, the polyphasic approach based on the different employed methodologies leads to a reliable final species identification. It is an important approach when food safety is the main subject, as is the case in the production of corn kernels.
4. Experimental Section
4.1. Corn Kernel Samples
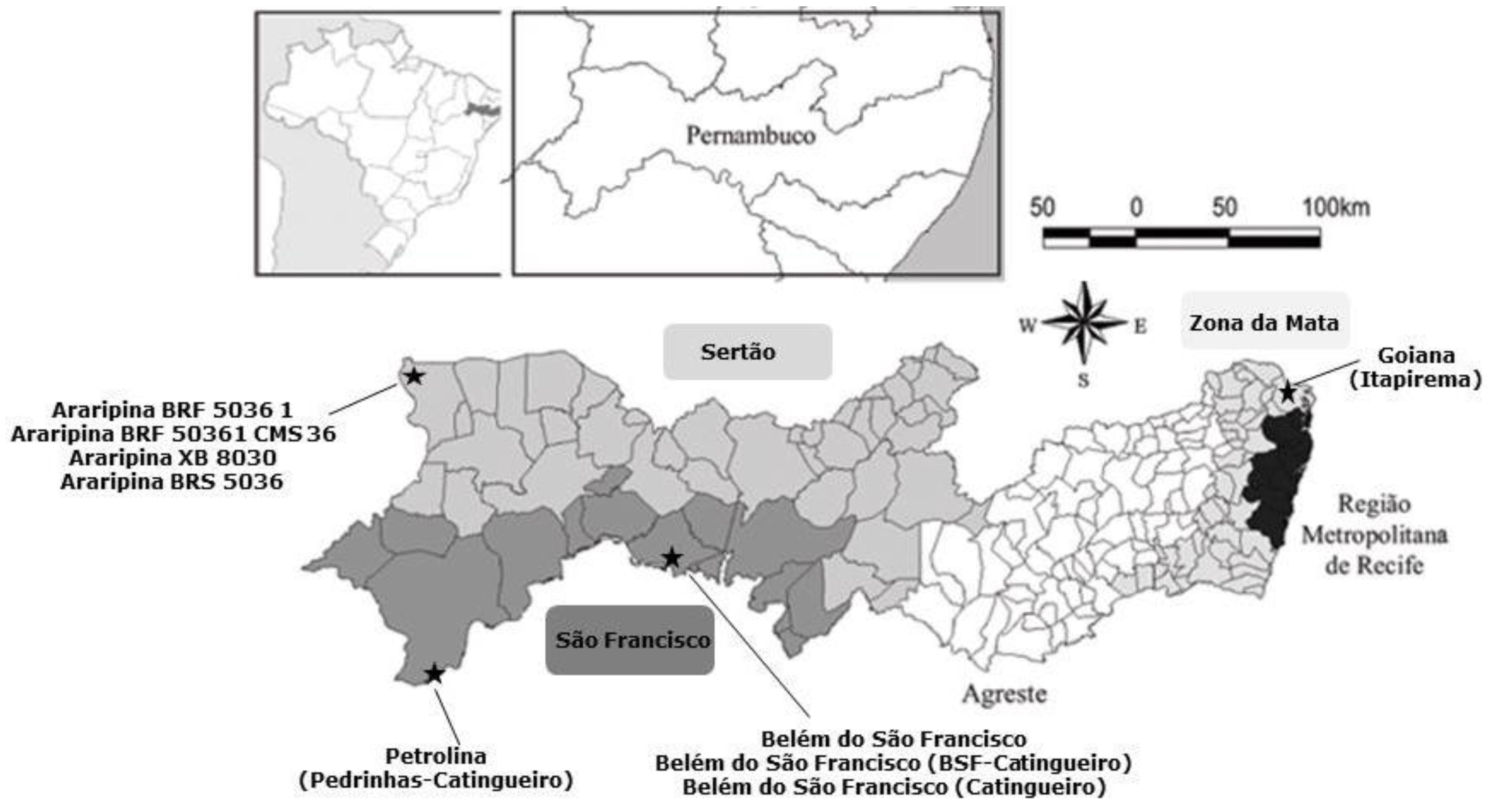
A total of 40 corn cobs were freshly harvested directly from the corn plant from July to August 2014 from different agriculture fields distributed among the following regions of Pernambuco State: São Francisco (15 corn cobs), Sertão (15 corn cobs) and Zona da Mata (10 corn cobs) (
Figure 5). Samples were provided by the Experimental Stations of the Agronomic Institute of Pernambuco-IPA and by local farmers.
4.2. Fungal Isolation and Identification
For the sample selection the phytosanitary of corn cobs was taken into consideration. Only visually heathy corn cobs free of fungal colonies, insects and lesions were selected. After selection, the corn cobs were stored in plastic bags, labelled, stored at −4 °C and transferred to the Laboratory of Fungal Molecular Genetics (Department of Mycology, Federal University of Pernambuco, Recife, Brazil).
A total of 200 hundred corn kernels randomly collected from the corn cobs harvested from the 3 different regions, as follow: São Francisco (15 corn cobs, 5 corn kernels per each cob), Sertão (15 corn cobs, 5 corn kernels per each cob) and Zona da Mata (10 corn cobs, 5 corn kernels per each cob). The corn kernels were surface-sterilised in 70% ethyl alcohol for 30 s, in 1.5% sodium hypochlorite for 5 min, rinsed thrice in sterile distilled water for 30 s, and subsequently dried with sterile paper towels. Corn kernel samples were further dried at room temperature under sterile conditions for 24 h. The kernels were then transferred to Petri dishes (10 kernels per plate) containing potato-dextrose-agar medium (PDA) supplemented with chloramphenicol (100 mg·L−1) and Dichloran-Glycerol agar-base (DG18, CM0729, Oxoid, Acumedia, Lansing, MI, USA) supplemented with chloramphenicol (100 mg·L−1). Samples were incubated at room temperature (25 ± 2 °C) for 5 days.
The numbers of fungal colonies were further determined and those fungal colonies indicative of
Fusarium sp. were sub-cultured in PDA and Dichloran-Glycerol (DG18) Agar Base plates and incubated for 5 days at 26 °C. Monospore cultures of
Fusarium species were subsequently evaluated by their macro- and micro-morphologies. Overall, macroscopic characteristics of each isolate, such as its colony colour and colony diameter, as well as the microscopic characteristics such as the shape and sizes of both micro- and macro-conidia, conidiophores, and phialides were assessed. Final fungal species identification followed the taxonomic keys and guides available for
Fusarium species [
29].
4.3. Mycelium Growth and DNA Extraction
Fusarium verticillioides isolates were inoculated on PDA plates and incubated at 25 °C for 7 days. Genomic DNA was extracted from the mycelium of each F. verticillioides isolate. Penicillium glabrum URM 3585 was used as negative control. The method for genomic DNA extraction was based on bead-beating at an agitation velocity of 5.0 m/s for 60 s in a microtube containing ca. 65 mg of mycelium and 700 μL of 2% CTAB buffer, pH 8.0 (previously heated to 65 °C).
Microtubes containing powdered fungal mycelium were incubated at 65 °C for 1 h and subsequently centrifuged at 12,000 rpm for 10 min at 24 °C. Each supernatant was transferred to a sterile microtube and then 650 μL of chloroform/isoamyl alcohol (24:1) solution was added. The solution was then centrifuged at 10,000 g and the aqueous phase was transferred to a sterile microtube, in which an equal volume of absolute isopropanol previously cooled at −20 °C was added.
In order to precipitate the nucleic acids, the final solution was incubated at −20 °C for 60 min. Samples were centrifuged at 10,000 g for 10 min and supernatants were discarded. Sediments were subsequently suspended and washed in 1 mL of 70% ethanol. Mixtures were centrifuged at 10,000 g for 5 min and supernatants were discarded. Samples were incubated at 37 °C up to the total evaporation of alcohol. The DNA samples were suspended with 50 μL of Tris-EDTA buffer (pH 8.0). Samples were kept at 4 °C for 24 h and subsequently stored at −20 °C up to their analysis.
4.4. Molecular Biology
In order to identify
F. verticillioides at species level, the primers VERT-1 (5′-GTCAGAATCCATGCCAGAACG-3′) and VERT-2 (5′-CACCCGCAAATCCATCAG-3′) were used. In addition, the detection of the
FUM1 gene was carried out by using the primers VERTF-1 (5′-GCGGGAATTCAAAAGTGGCC-3′) and VERTF-2 (5′-GAGGGCGCGAAACGGATCGG-3′), as previously described elsewhere [
37].
The amplification reactions were undertaken in a final volume of 25 µL, containing: 20 mM Tris–HCl buffer (pH 8.4), 50 mM KCl, 0.75 mM MgCl
2, 2 mM dNTP, 20 pmol of the primer, 3 U/μL of
Taq DNA polymerase, and 25 ng of DNA [
12]. The Techne TC-512 thermocycler with the following programmed cycles was used: 94 °C for 1 min for initial denaturation, followed by 35 cycles of denaturation for 1 min at 94 °C, annealing at 60 °C for 1 min, extension at 72 °C for 1 min, and a final extension at 72 °C for 5 min. The amplified products and the 1 Kb plus DNA ladder (Invitrogen Life Tecnologies, Carlsbad, CA, USA) were stained with
GelRedTM (Biotium, Hayward, CA, USA), separated by electrophoresis (Hoefer, Holliston, MA, USA) on 1.0% agarose gel at 3 V·cm
−1 in TAE (1×) (pH 8.0) running buffer and visualised in an ultraviolet light transilluminator (Vilber Lourmat, Suarlée, Belgium). Images were recorded using a Sony digital camera (7.2 effective mega pixels) (Sony, Toquio, Japan).
4.5. Analyses of Genetic Variability
In order to evaluate the genetic variability of the
F.
verticillioides strains, PCR reactions using the ISSR primers (GTG)
5 [
38] and (GACA)
4 [
39] were used. Amplification reactions were performed in a final volume of 25 µL using the following contents: 20 mM Tris-HCl buffer (pH 8.4), 50 mM KCl, 0.75 mM MgCl
2, 0.25 mM dNTP, 0.25 mM of the primer, 0.4 U of
Taq DNA polymerase (Operon Technologies, Alameda, CA, USA), and 25 ng of DNA.
Amplification reactions were performed in a Techne TC-512 thermocycler (Analitica, São Paulo, Brazil) with the following programmed cycles: an initial denaturation step at 93 °C for 5 min, 40 cycles of 20 s at 93 °C, 45 s at 55 °C and 90 s at 72 °C, followed by a final extension at 72 °C for 6 min. The amplified products and the 1 Kb plus ladder DNA (Invitrogen Life Tecnologies) were stained with GelRedTM, separated by electrophoresis in 1% agarose gel at 4 V′cm−1 in the running buffer TAE (1×) (pH 8.0). The gel was visualised after electrophoresis by using an ultraviolet transilluminator, and registered using a Sony digital camera (7.2 effective mega pixels).
4.6. Statistical Analyses NTSYS-PC
The data obtained from the PCR amplifications using (GTG)
5 and (GACA)
4 primers were analysed using the Numerical Taxonomy System of Multivariaty Programs—NTSYS-PC [
40]. The data were introduced in the form of binary variables, with the number 1 meaning the presence of a band, and the number 0 (zero) its absence. A matrix of similarity was then constructed by using Jaccard coefficient (J) [
41]. The matrix of similarity was used to construct a dendrogram based on the Unweighted Pair Group Method with Arithmetical Average (UPGMA).
4.7. Fungal Identification by MALDI-TOF MS
Seven-day-old spores of each Fusarium suspended in 0.5 mL of 0.2% agar were used for inoculation on a single-point 6 cm PDA plate. All isolates were incubated for 3 days at 25 °C. Approximately 1 μg of mycelium or mycelium/spores mixture from the growing edge colony was transferred directly from the culture plate to the 48-well MALDI flex target plate (Flexi-Mass™, Shimadzu Biotech, Manchester, UK). Immediately, 0.5 μL matrix solution (75 mg/mL 2,5-dihydroxybenzoic acid [DHB] in ethanol/water/acetonitrile (1:1:1) with 0.03% trifluoroacetic acid [TFA]) was added and mixed gently. Escherichia coli DH5α was obtained from the Public Portuguese Culture Collection Micoteca da Universidade do Minho-MUM and its cells were used for in situ extraction of ribosomal proteins, which were used as standard for the MALDI-TOF MS external calibration. Cells of E. coli DH5α were grown on Luria-Bertani medium agar (LB; 10 g∙L−1 bacto-tryptone, 5 g∙L−1 bactoyeast extract, 10 g∙L−1 NaCl) at 30 °C for 20 h. About 1 μg cellular material from a single E. coli DH5α colony was transferred from the LB plate to the MALDI flex plate and the matrix solution was applied as described above for the fungal analysis. All sample plates were air dried at room temperature. Each sample was spotted in duplicate to test reproducibility. During the analyses, all solutions were prepared daily and stored at 5 °C.
The analyses were performed on an Axima LNR MALDI-TOF MS system (Kratos Analytical, Shimadzu, Manchester, UK) equipped with a nitrogen laser (337 nm), where the laser intensity was set just above the threshold for ion production. Twelve defined ribosomal proteins of intact E. coli DH5α cells (4365.4, 5096.8, 5381.4, 6241.4, 6255.4, 6316.2, 6411.6, 6856.1, 7158.8, 7274.5, 7872.1, 9742 and 12,227.3 Da) were used as external calibrants. The mass spectra based on the mass range from 2 to 20 kDa were recorded using the linear mode with a delay of 104 ns and an acceleration voltage of 20 kV. The final spectra were generated by summing 20 laser shots accumulated per profile and 50 profiles produced per sample, which led to 1000 laser shots per summed spectrum.
The resulting peak lists were exported to the SARAMIS™ software package (Spectral Archiving and Microbial Identification System, AnagnosTec, Version 2010, Potsdam, Germany, 2010) where the final microbial identification was achieved. In SARAMIS™, peak lists of individual samples were compared on database generating a ranked list of matching spectra. This software uses a point system based on peak list with mass signals weighted according to their specificity. The similarity between individual spectra is expressed as the relative or absolute number of matching mass signals after subjecting the data to a single link agglomerative clustering algorithm. Microbial identifications by the SARAMIS™ package are based on the presence or absence of each peak in the spectra. A dendrogram of proteomic profile proximity among isolates was created using SARAMIS™ package.
4.8. Detection of Fumonisins by MALDI-TOF MS/MS
Samples were prepared according the previous fumonisins B1 and B2 extraction method described by Ono
et al. [
42] with some adaptation. Briefly, all of the
F. verticillioides isolates were grown on a single-point on 6 cm PDA plates at 28 °C for 7 days. Each isolate (10
6 conidia/mL) was then inoculated onto a 6 cm Petri dish containing ground corn moistened with distilled water (1 g/mL
v/
v), and autoclaved two times for 30 min at 121 °C. The cultures were incubated at 25 °C for 14 days. Three plugs of each culture samples were then extracted with 1 mL methanol-water (3:1,
v/
v). After 10 min incubation at room temperature, the suspension was shaken at 150 rpm for 30 min and centrifuged at 4500
g for 10 min.
Supernatants were then transferred to amber flasks and liquid phase was evaporated at room temperature overnight. Residues of each sample were then suspended in 0.5 mL methanol and 1 µL of each sample was mixed on a paraffin film surface with 2 µL α-cyano-4-hydroxycinnamic acid-CHCA (Fluka, Buchs, Switzerland) saturated in a solution with 33% ethanol, 33% acetonitrile, 31% H2O and 3% TFA. Each suspension (1 µL) was spotted onto a MALDI-TOF stainless plate (Bruker Daltonics, Billerica, MA, USA). Samples were air-dried at room temperature prior to spectral acquisition. In order to test their reproducibility samples were analysed in triplicate. In case of discordant results, analysis was repeated for at least 2 additional replicates.
During the sample preparation, all solutions were prepared and stored at 5 °C. Samples were finally analysed in a MALDI-TOF MS/MS Autoflex III (Bruker Daltonics, Billerica, MA, USA), equipped with a Nd:YAG laser at 355 nm. The spectral acquisition was assessed in the reflector positive mode, with an acceleration voltage of 19 kV under a mass range between m/z 500–4480. The external calibration was performed by using the Peptide Calibration Standard II (Bruker Daltonics).



 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}