
Advanced Oxidation Protein Products-Modified Albumin Induces Differentiation of RAW264.7 Macrophages into Dendritic-Like Cells Which Is Modulated by Cell Surface Thiols


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
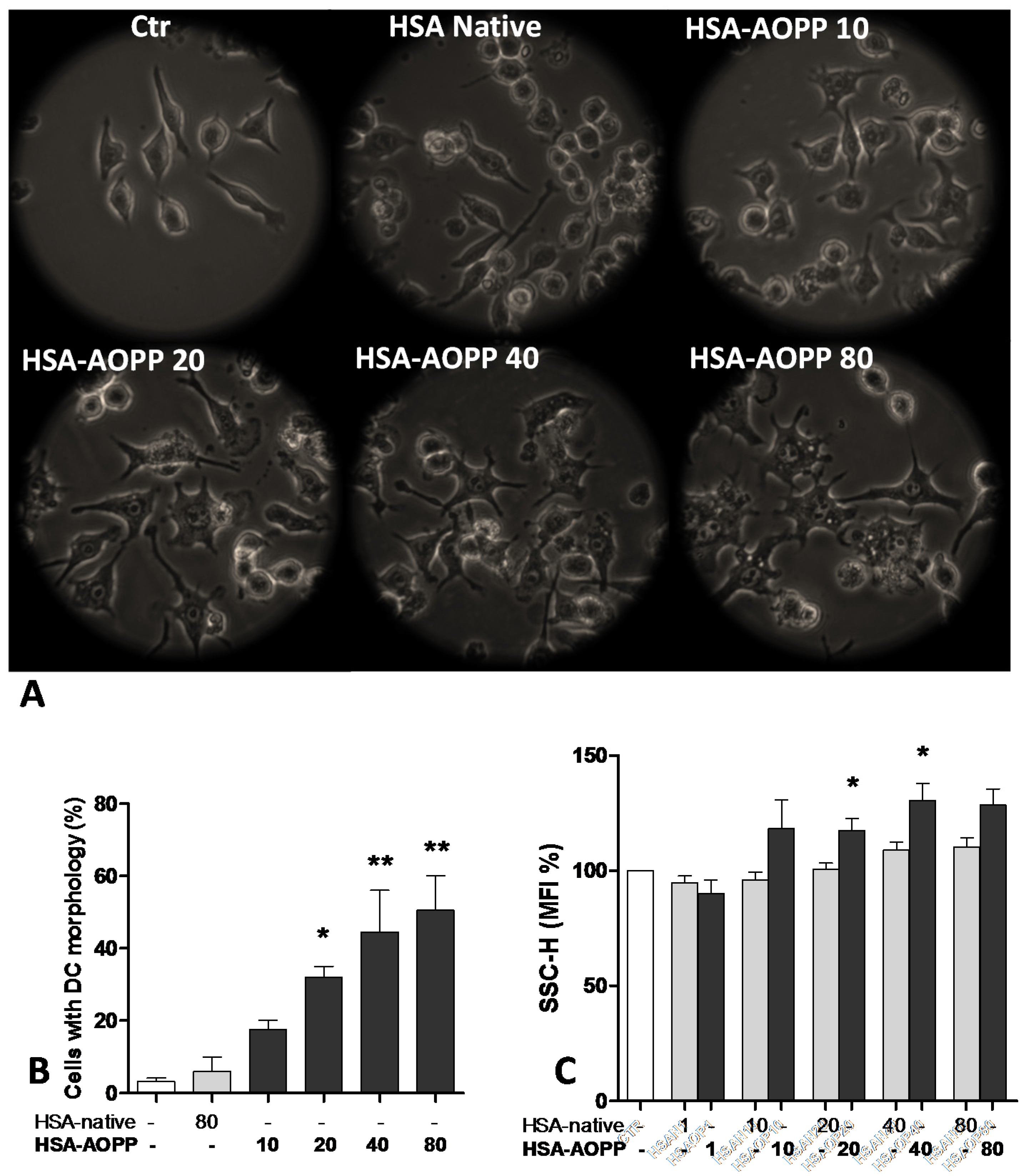
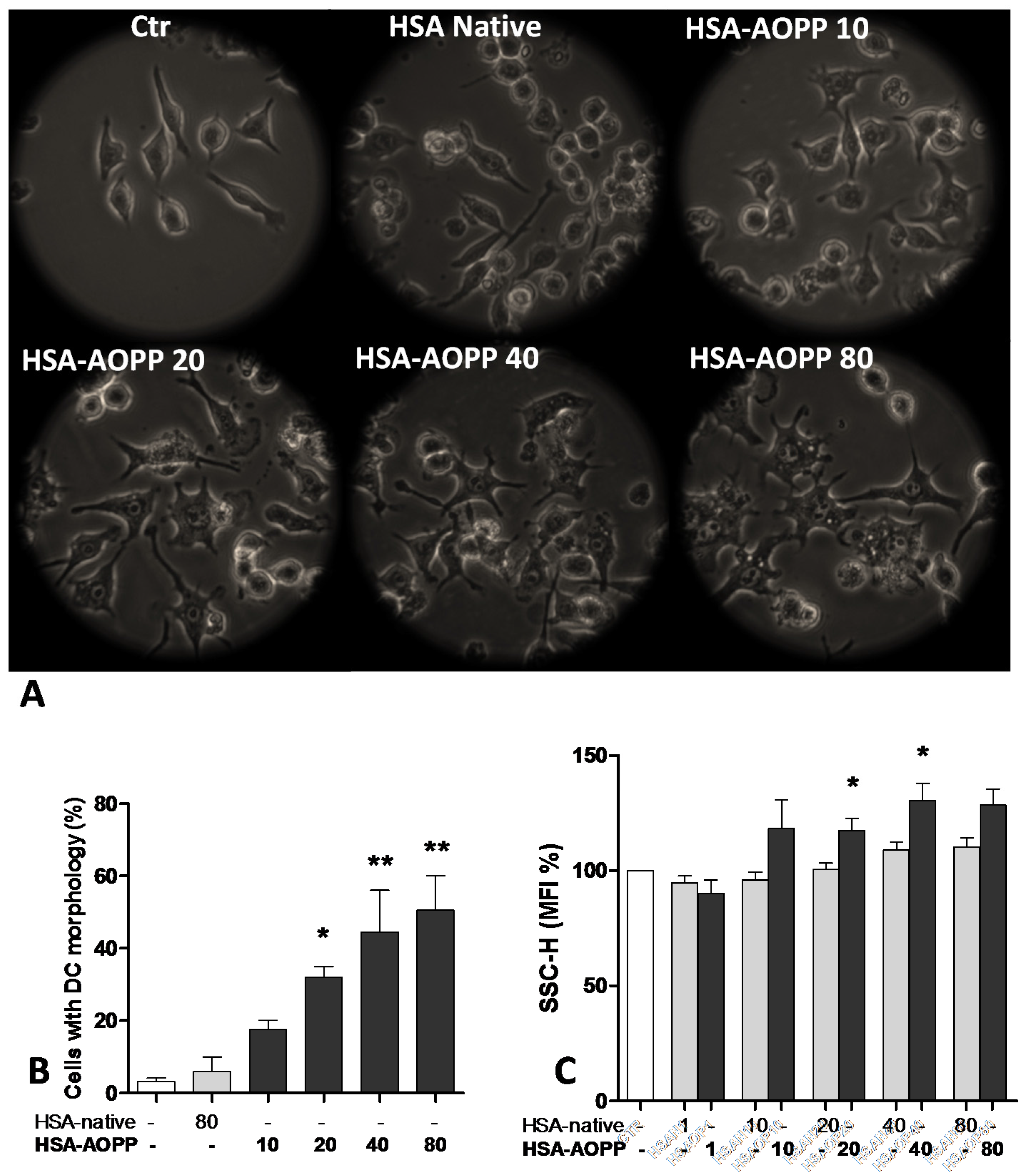
2.1. AOPP Induced Morphologic Changes in RAW264.7 Cells toward DC-Like Cells
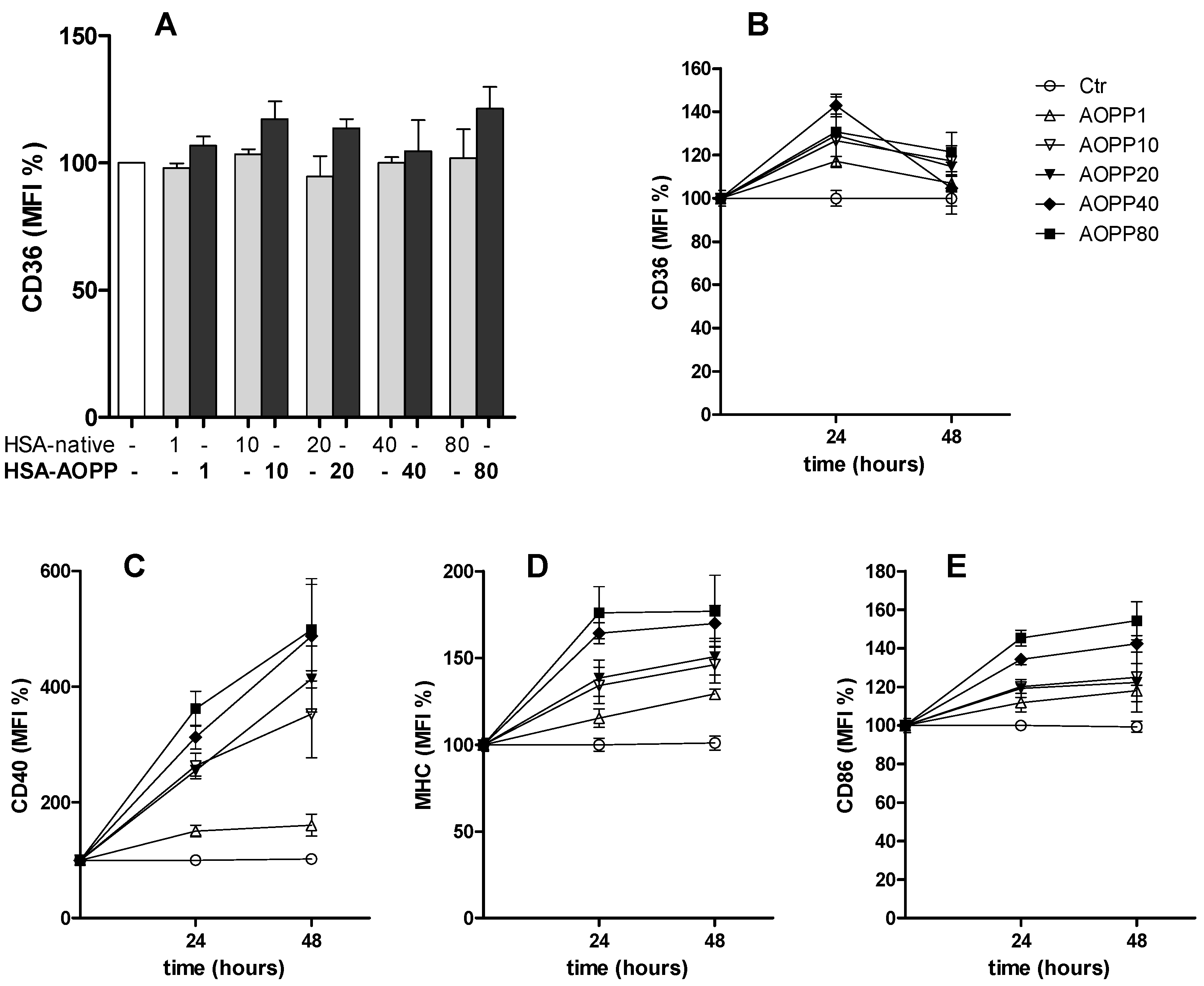
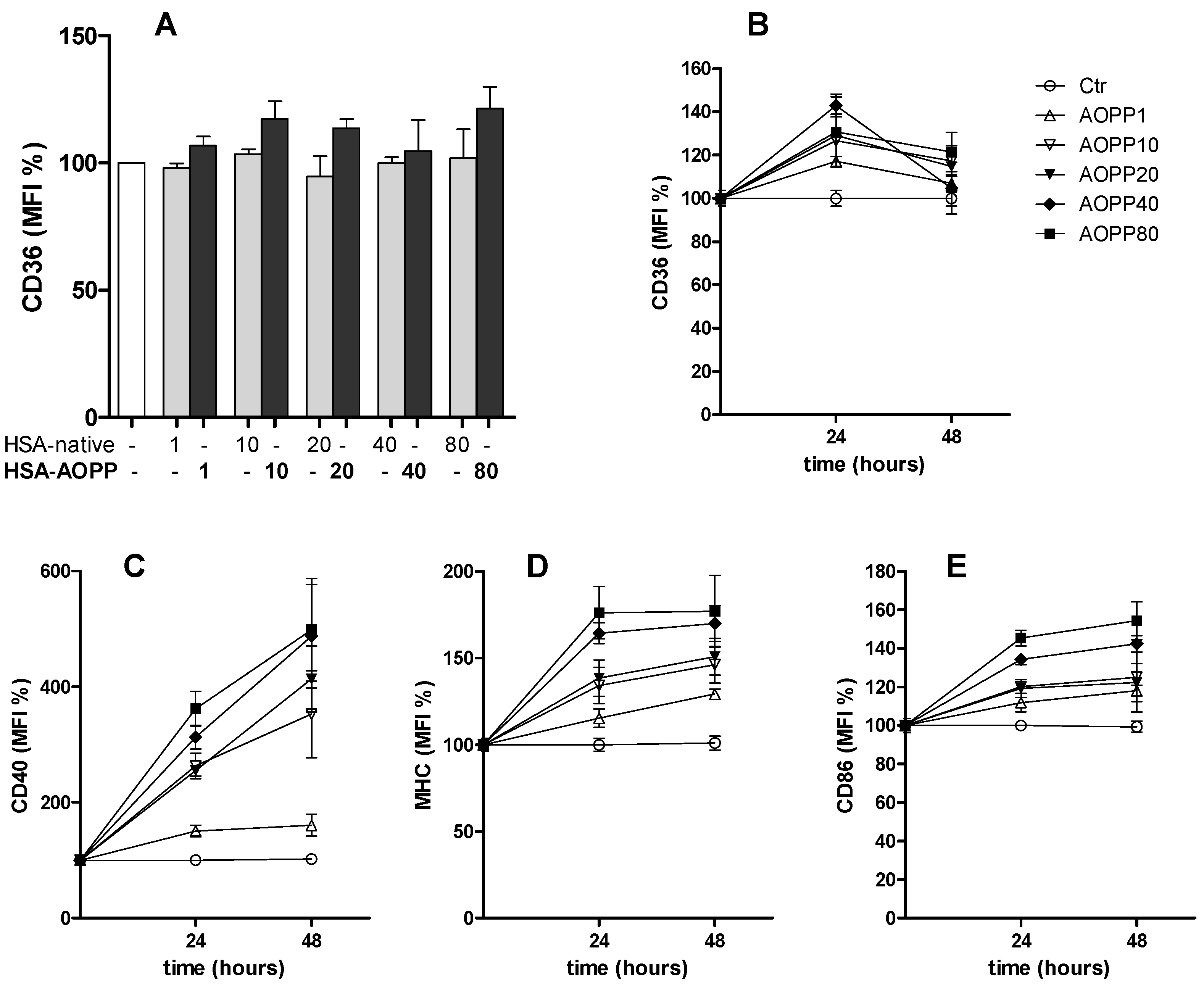
2.2. CD36 Expression in RAW264.7 Cells and Time Course of Surface DC Markers upon Treatment with HSA-AOPP
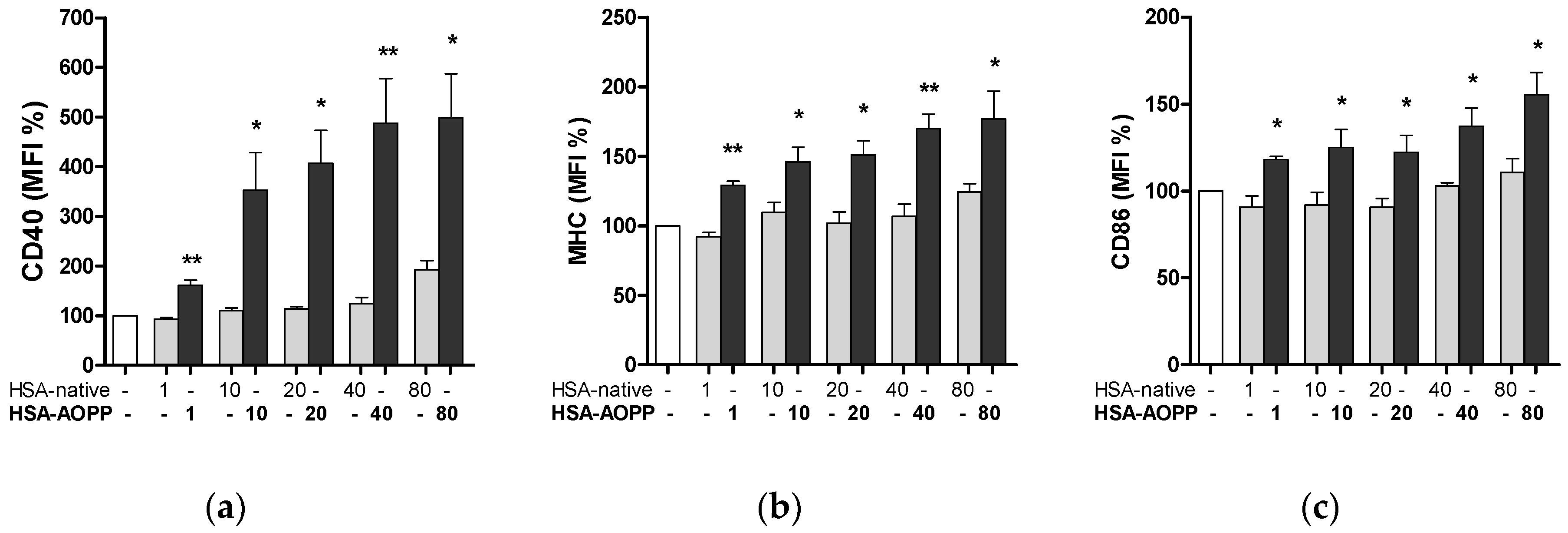
2.3. HSA-AOPP Induced Phenotypic DC Markers Expression in RAW264.7 Cells. Flow Cytometry of Phenotypic Parameters
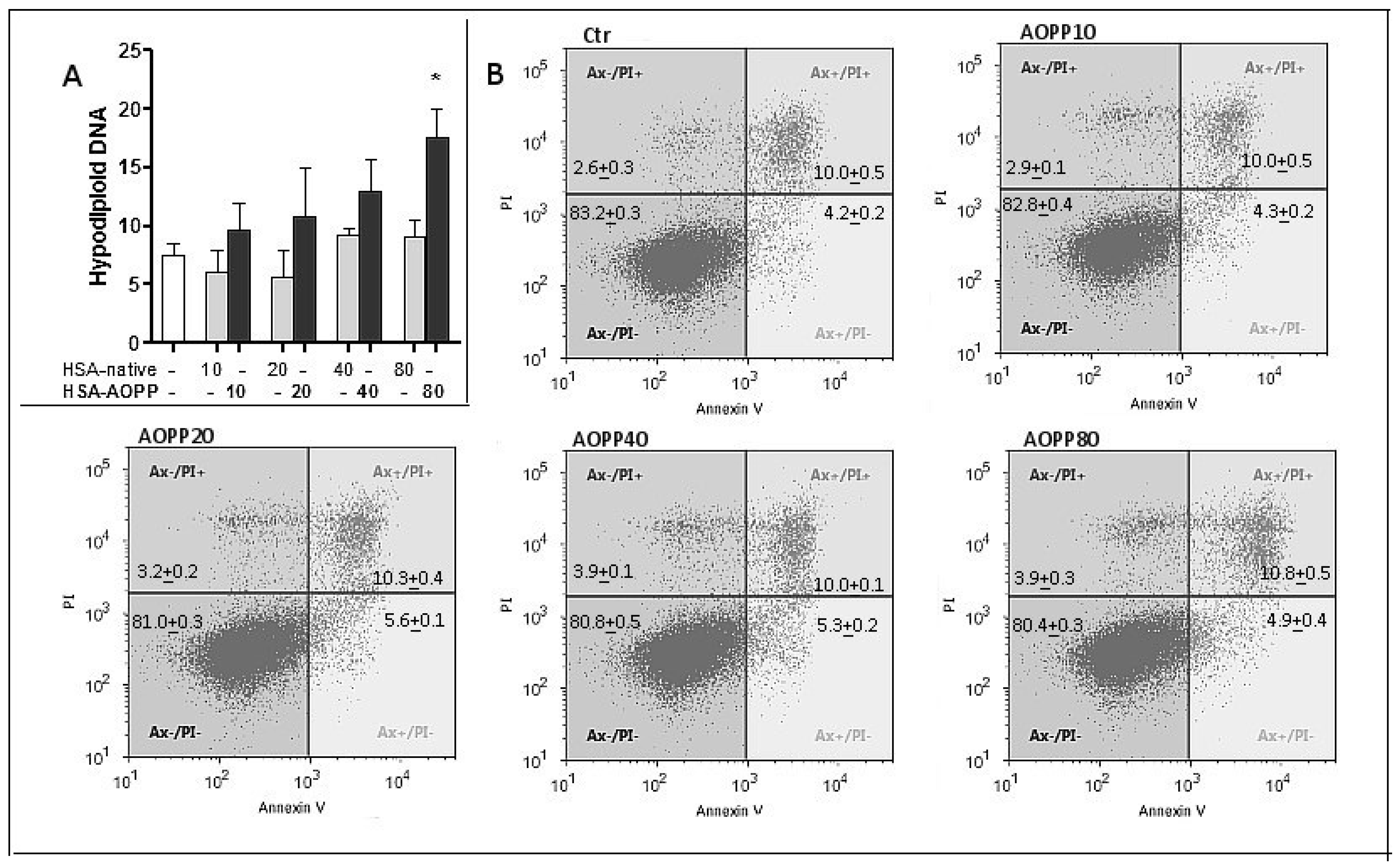
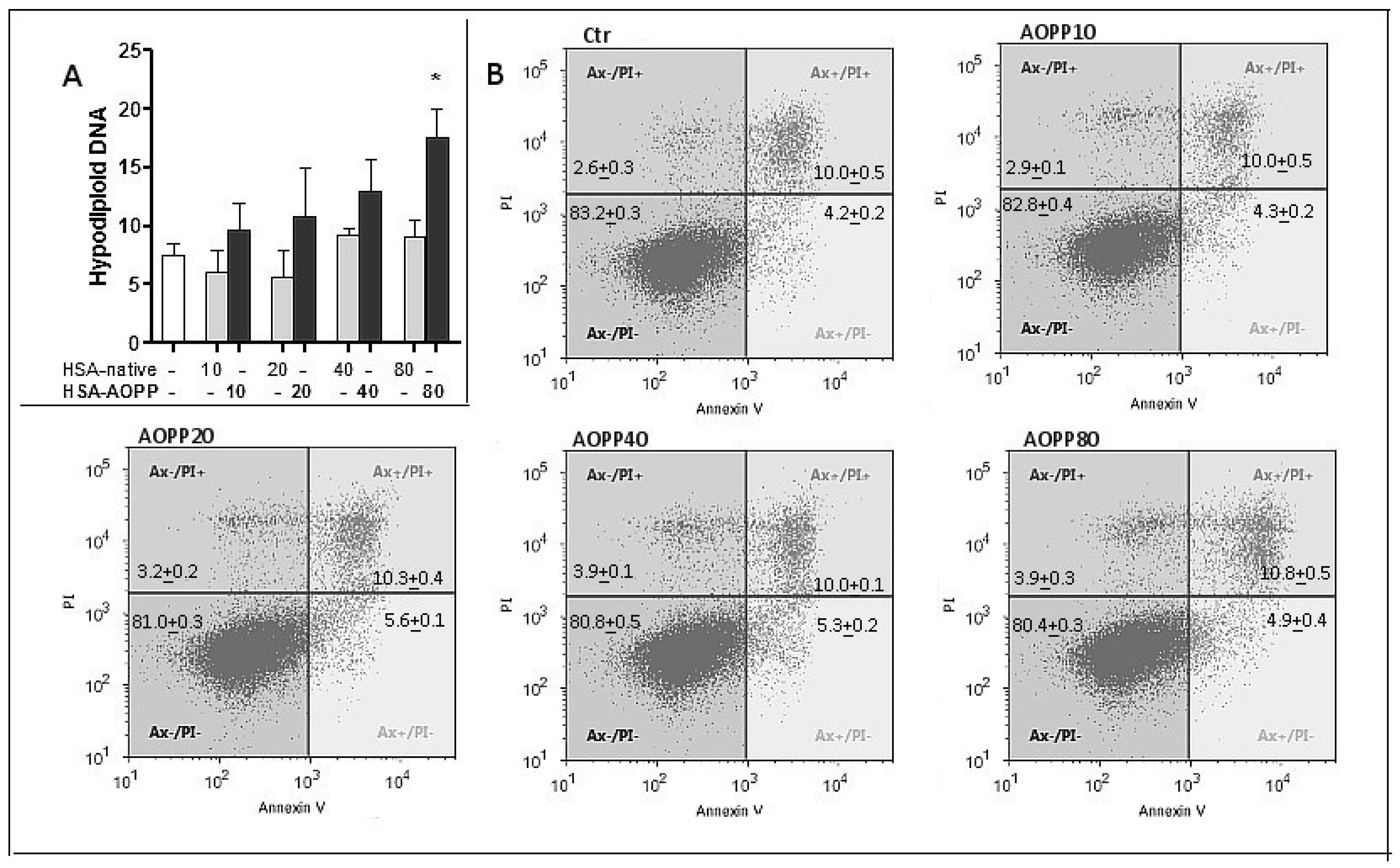
2.4. Evaluation of Cell Viability
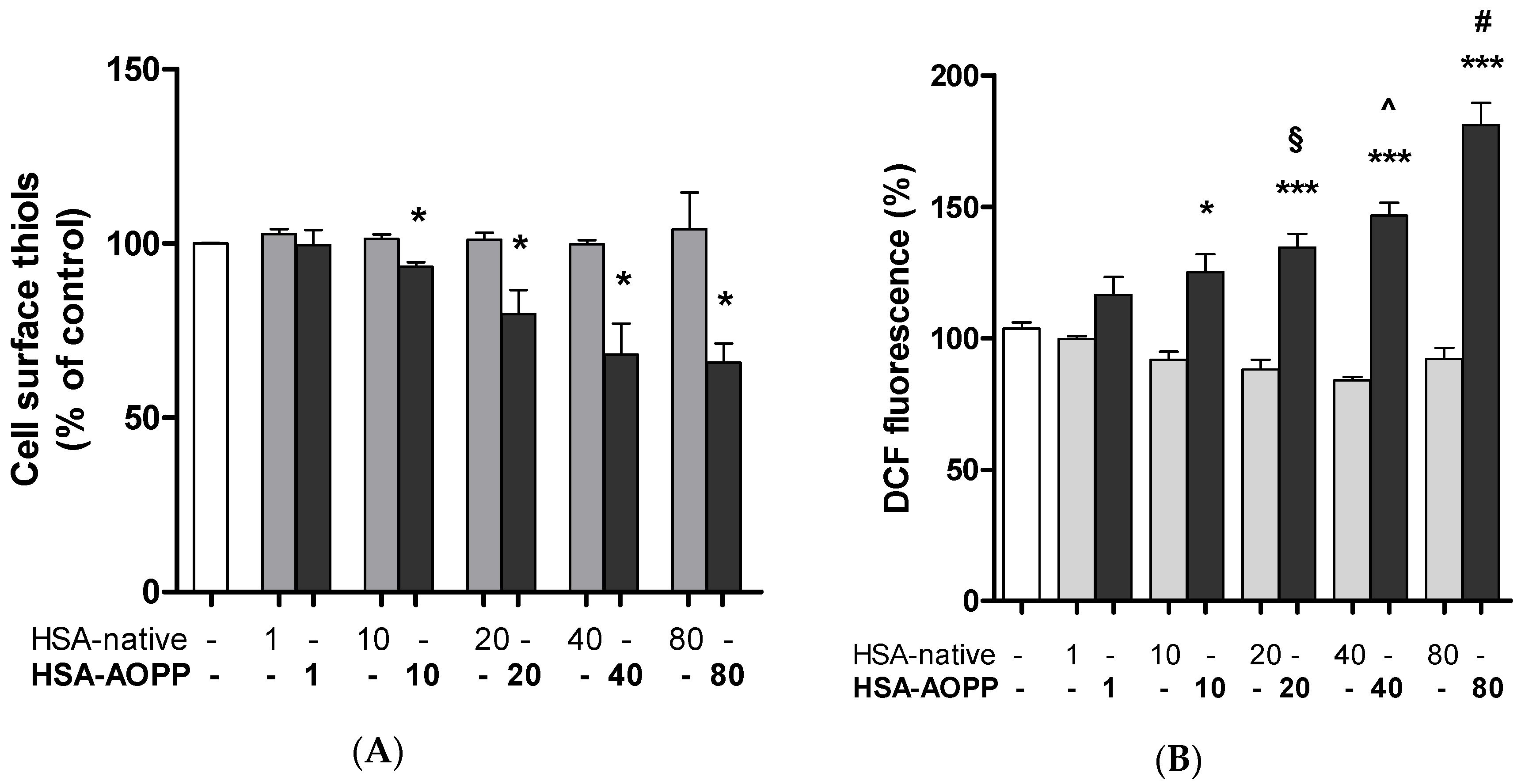
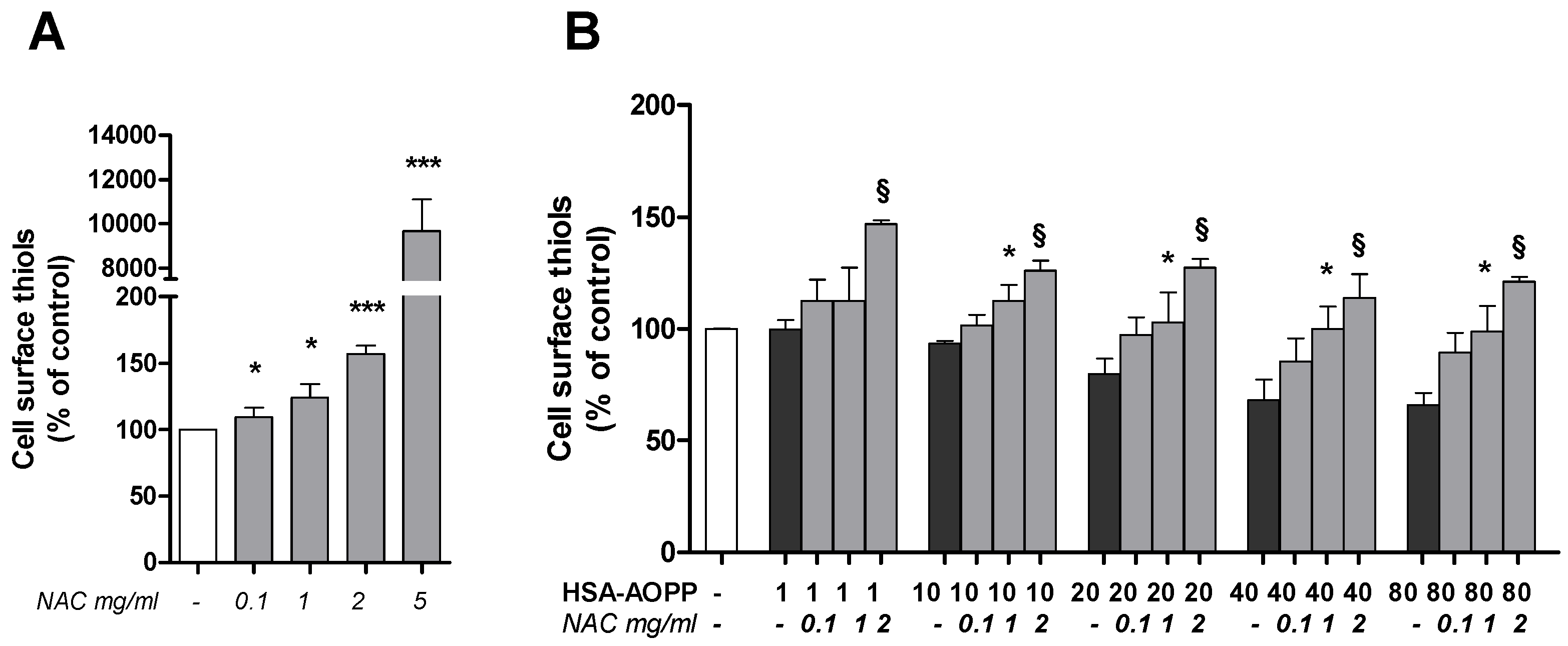
2.5. Cell Surface Thiol Groups and Intracellular ROS Production Are Modulated by HSA-AOPP
2.6. N-Acetylcysteine (NAC) Pre-Treatment Enhanced the Surface Thiols and Prevented Their AOPP-Dependent Decrease
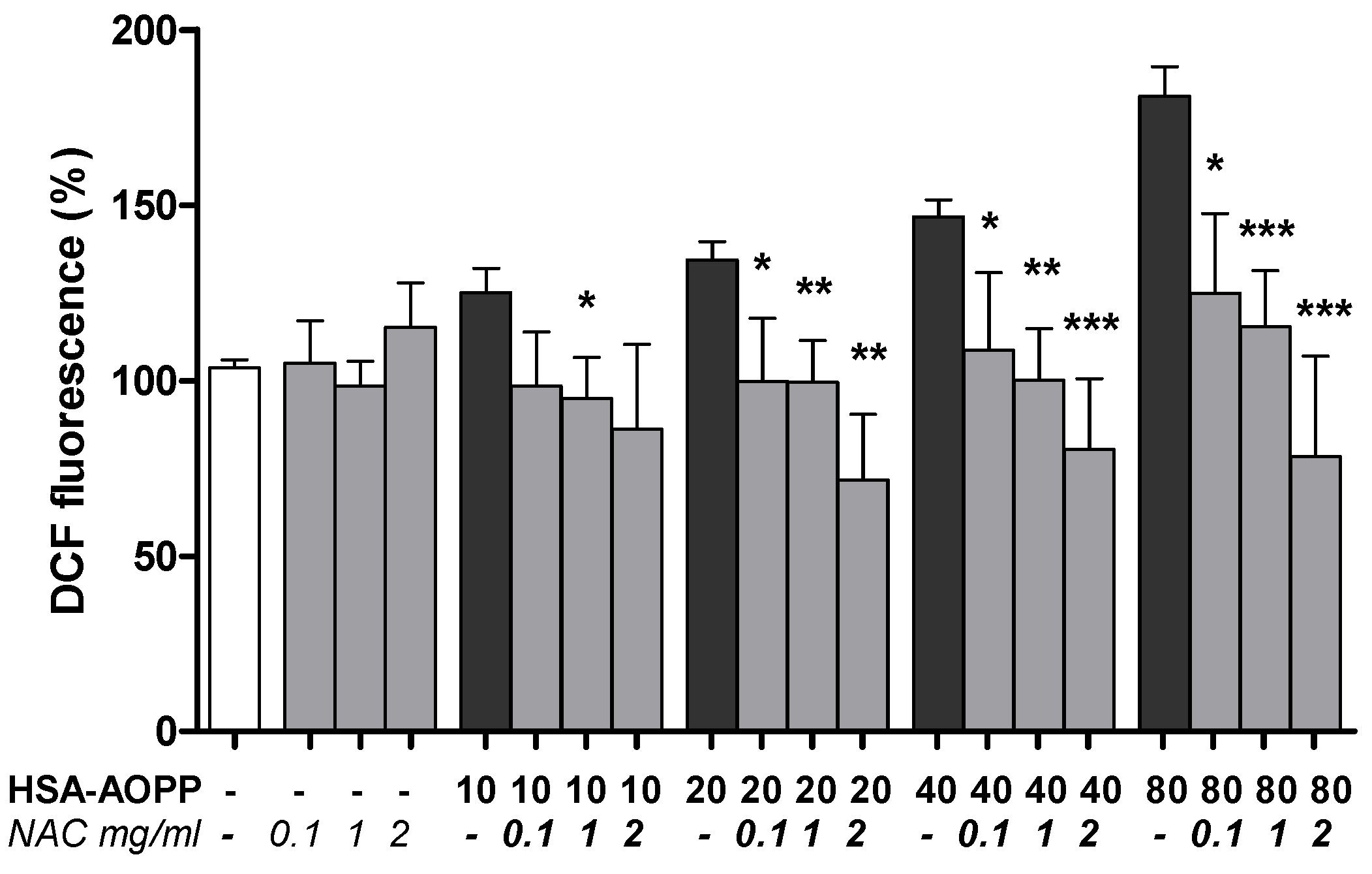
2.7. NAC Pre-Treatment Reduced ROS Production
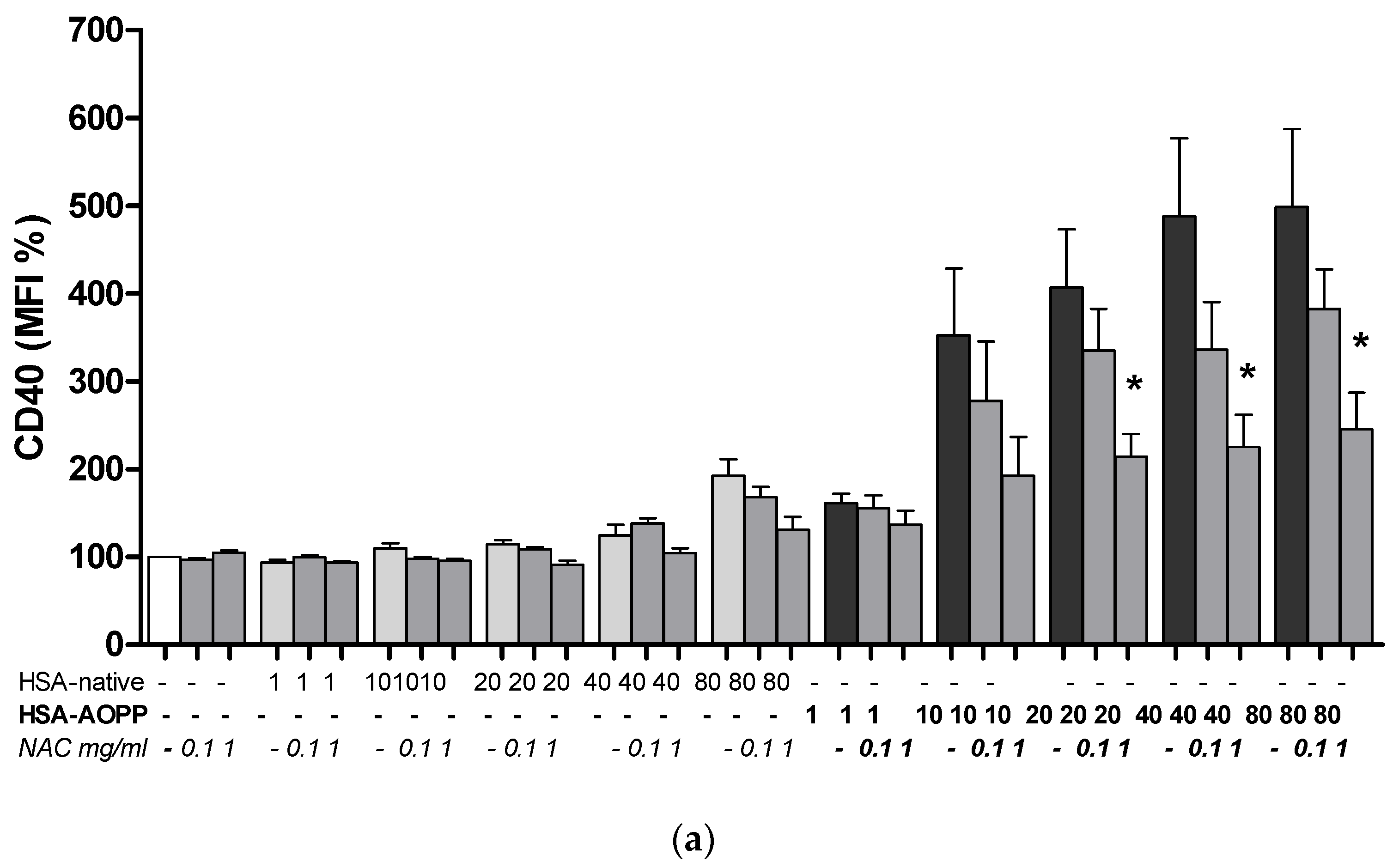
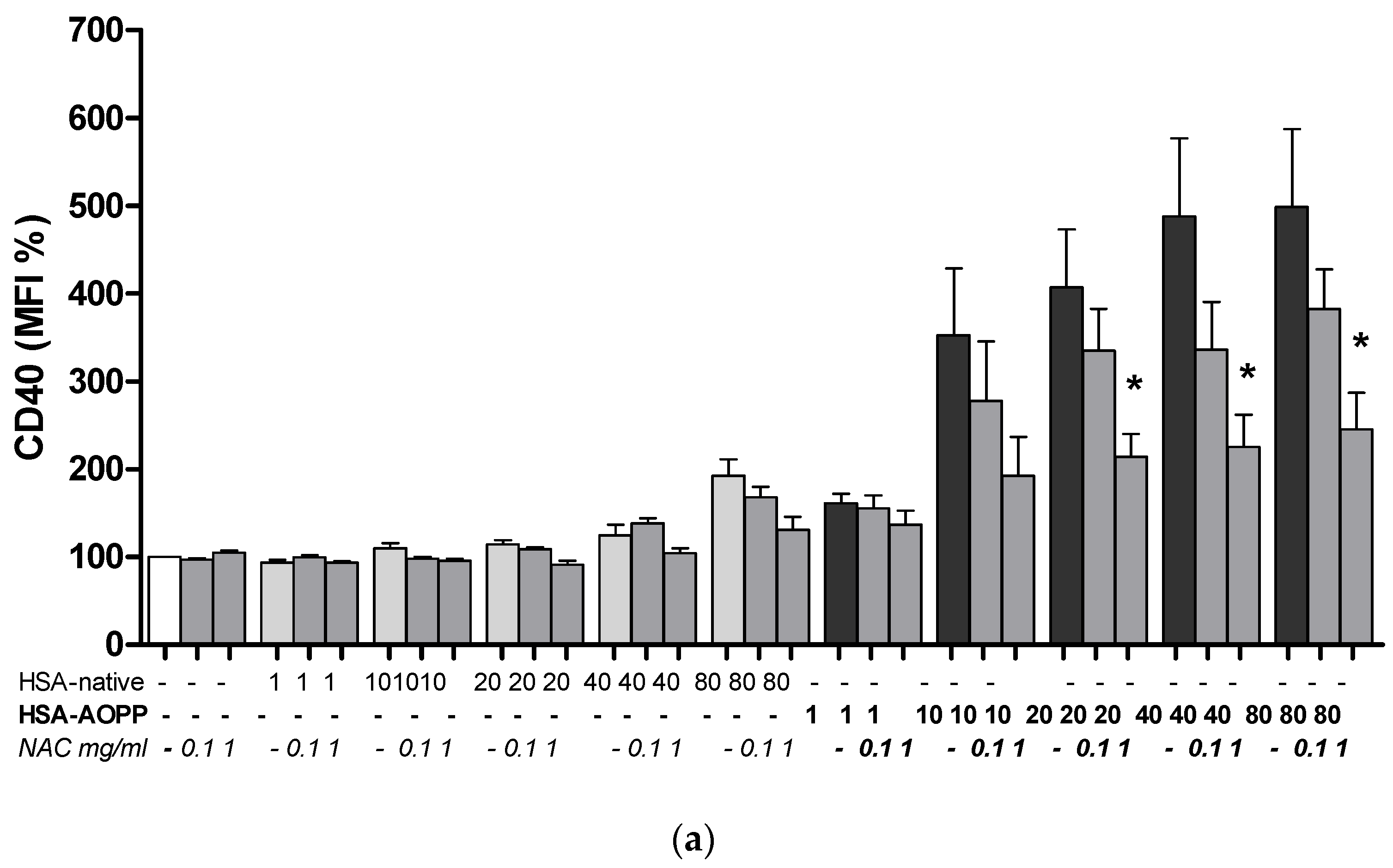
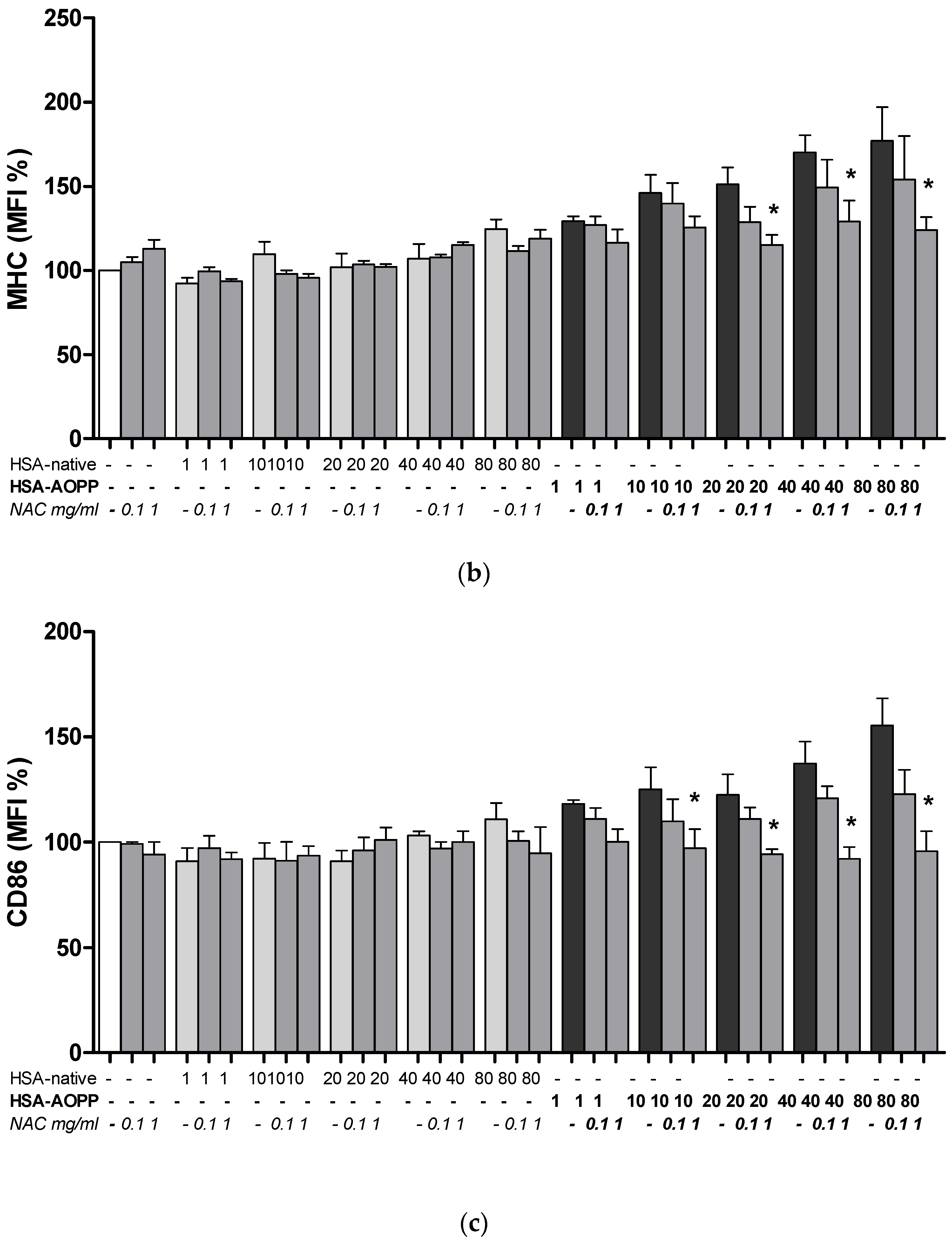
2.8. The Effect of NAC Treatment on Cell Surface Markers
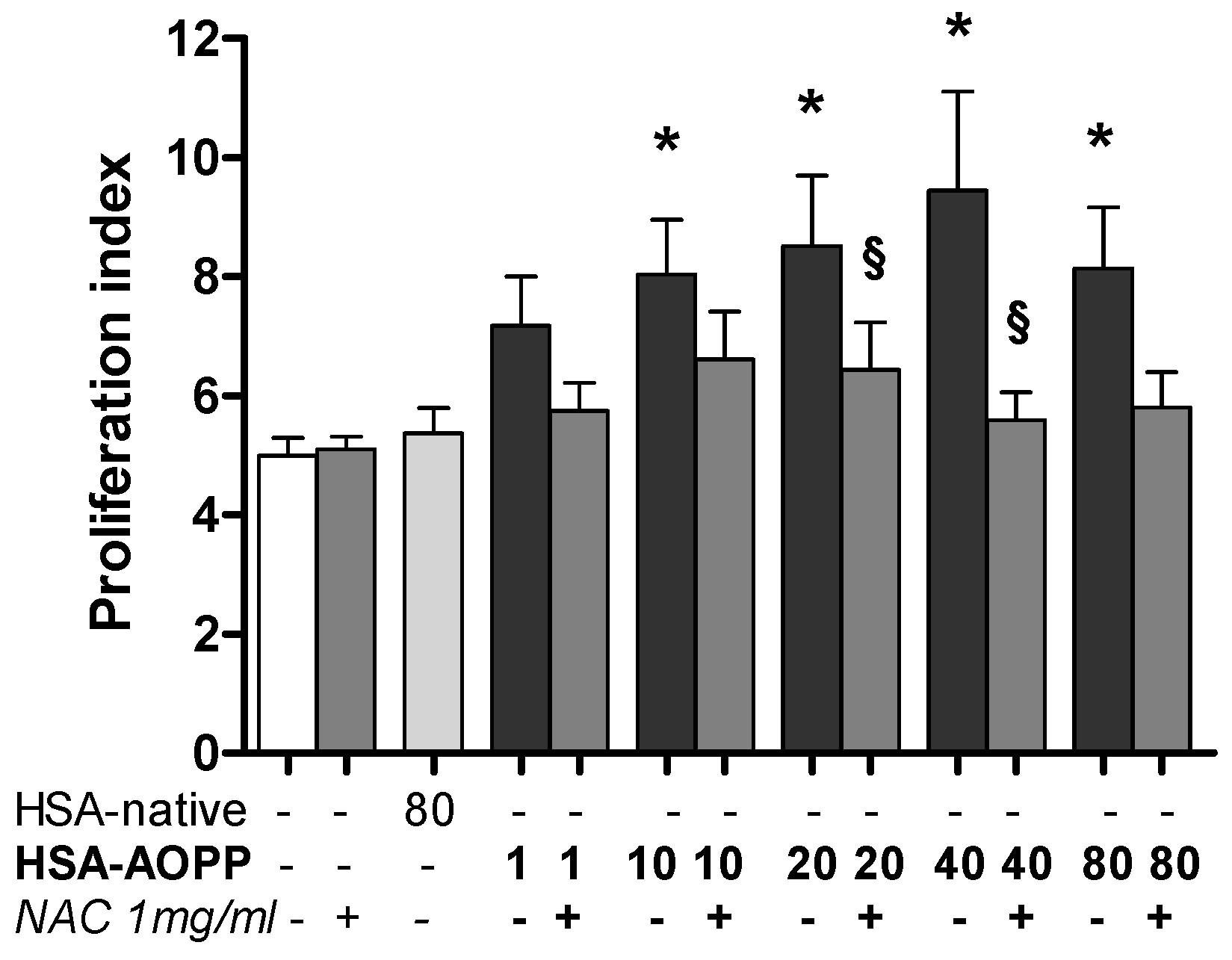
2.9. Dendritic Cell Functional Assay. MLR for T-Cell Proliferation Induced by HSA-AOPP Treated RAW264.7 Cells
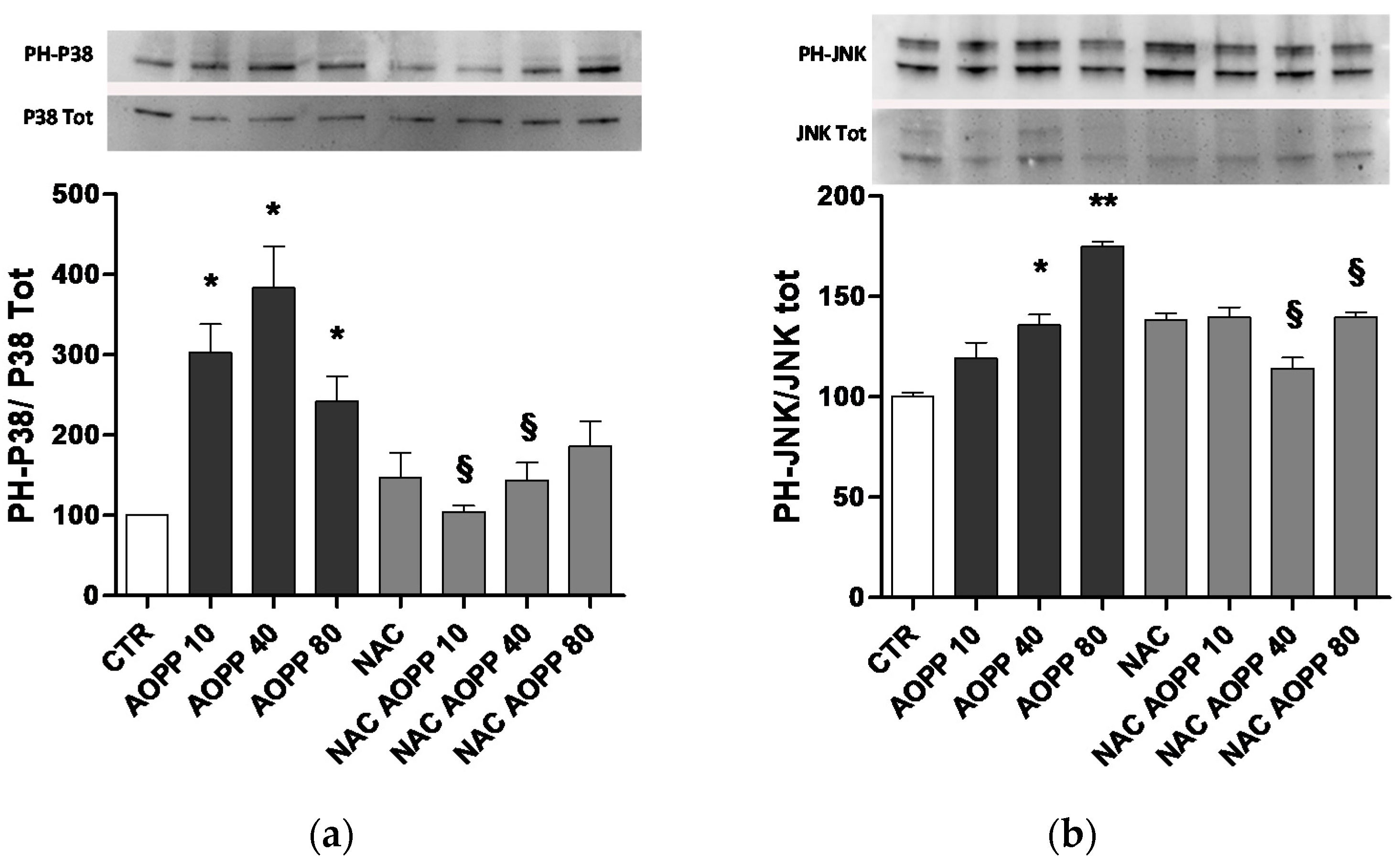
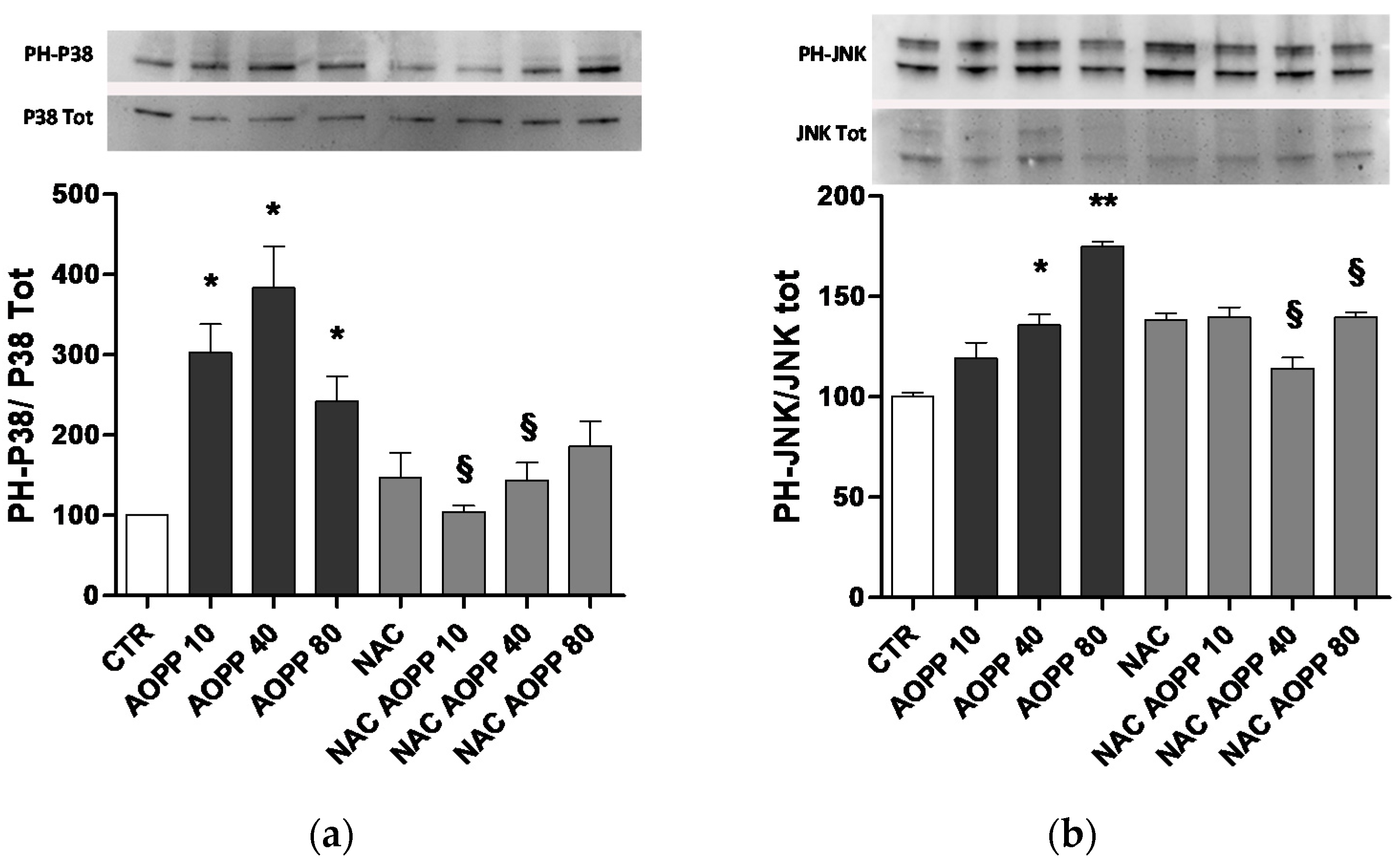
2.10. p38 MAPKinase and JNK Are Affected by AOPP Treatment of RAW264.7 Cells
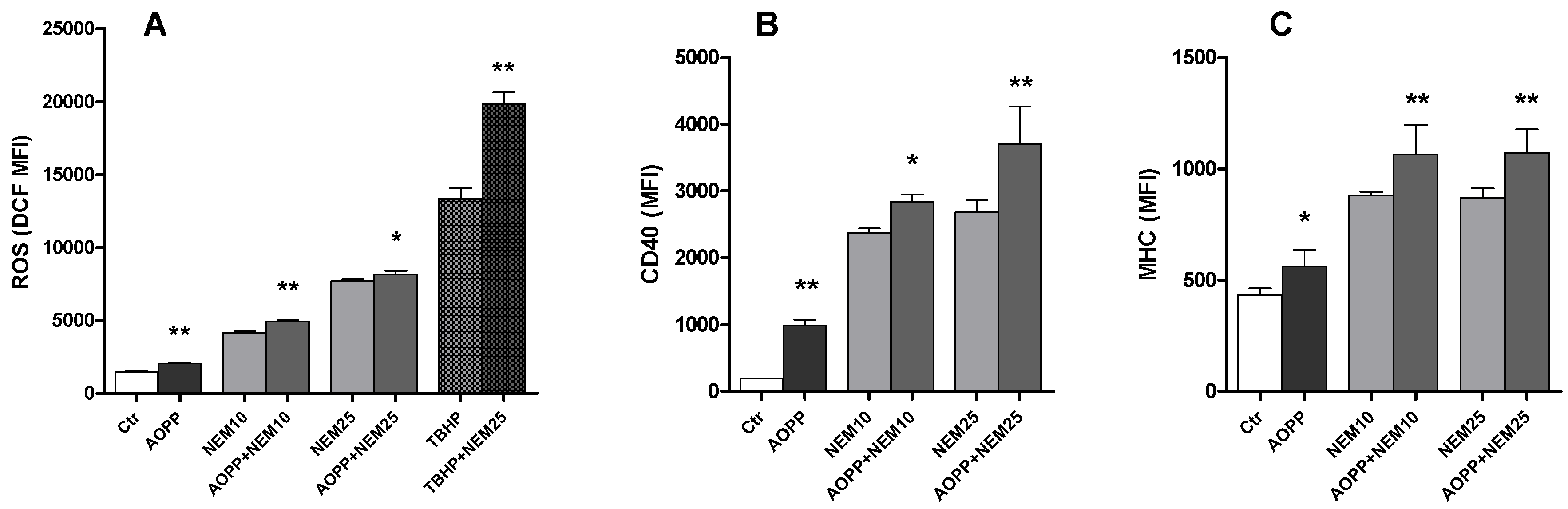
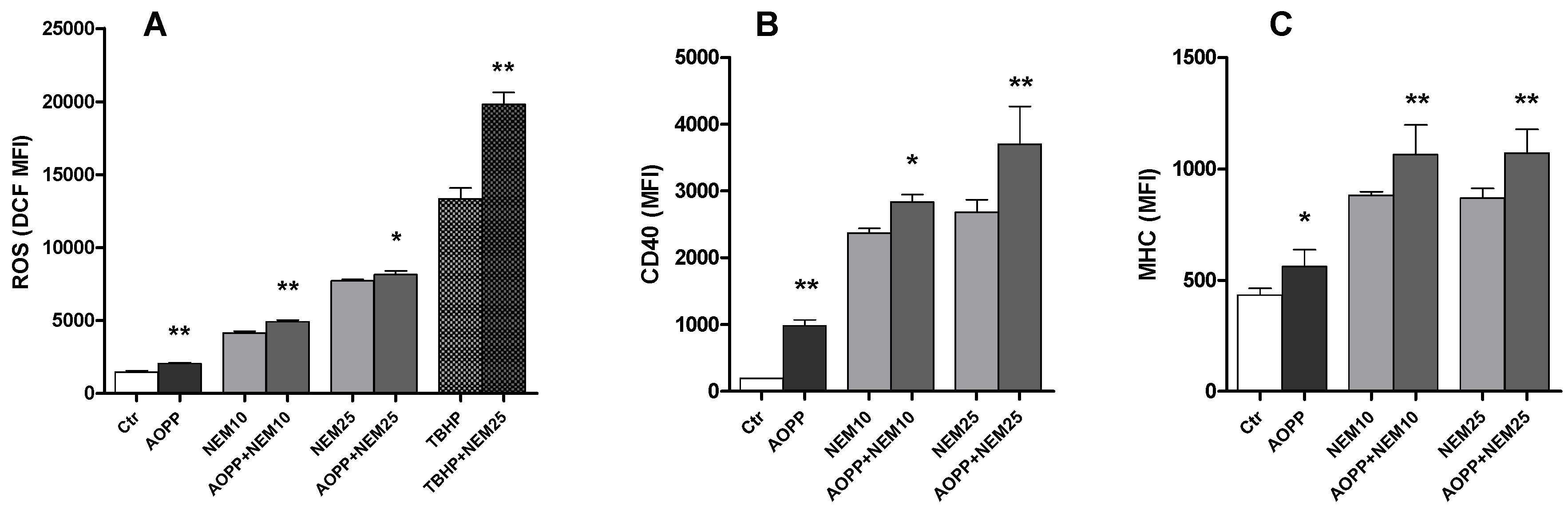
2.11. Effects of N-Ethylmaleimide, a Thiol Modifier, on RAW264.7 Cell ROS Production and DC Markers
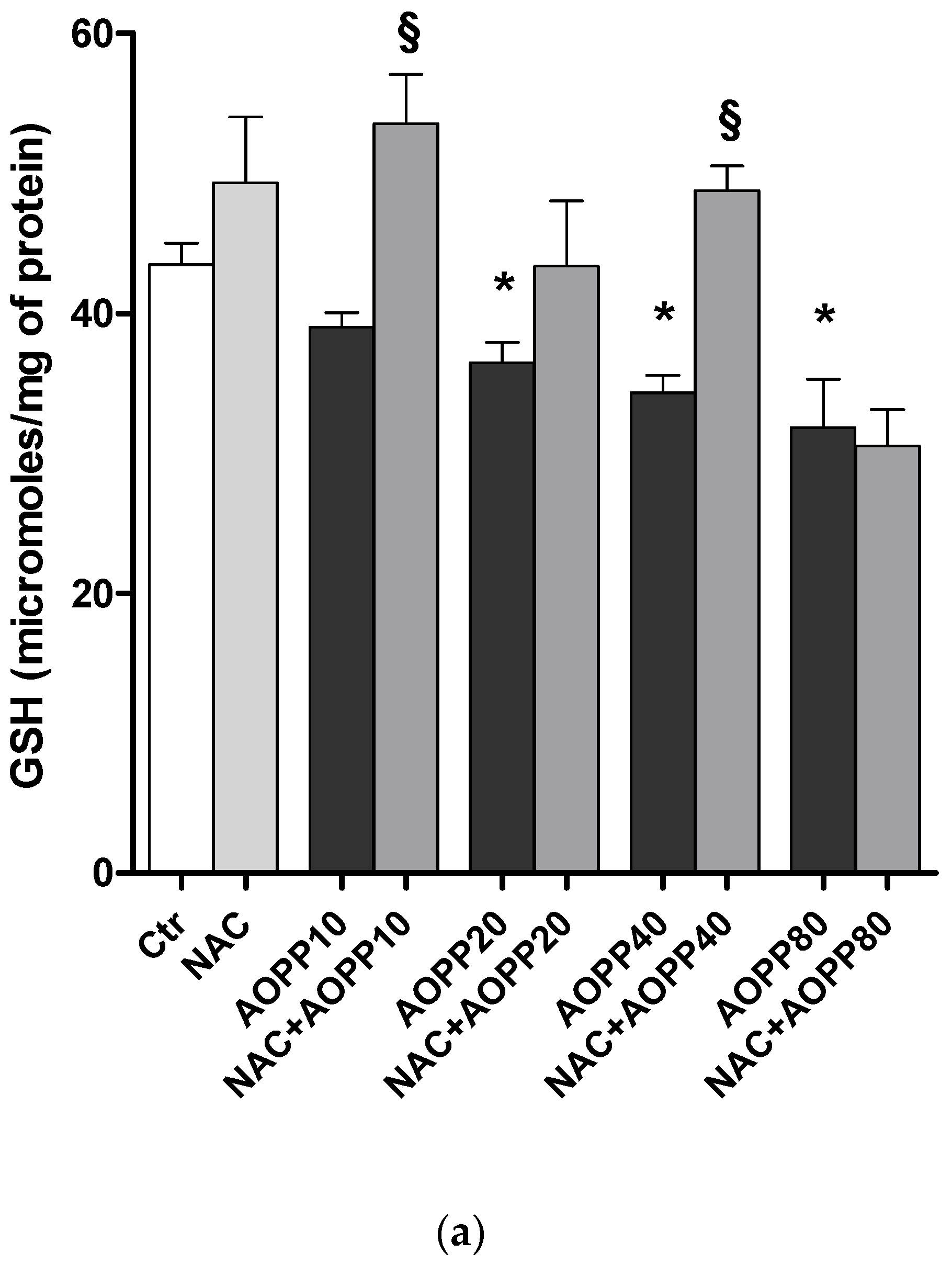
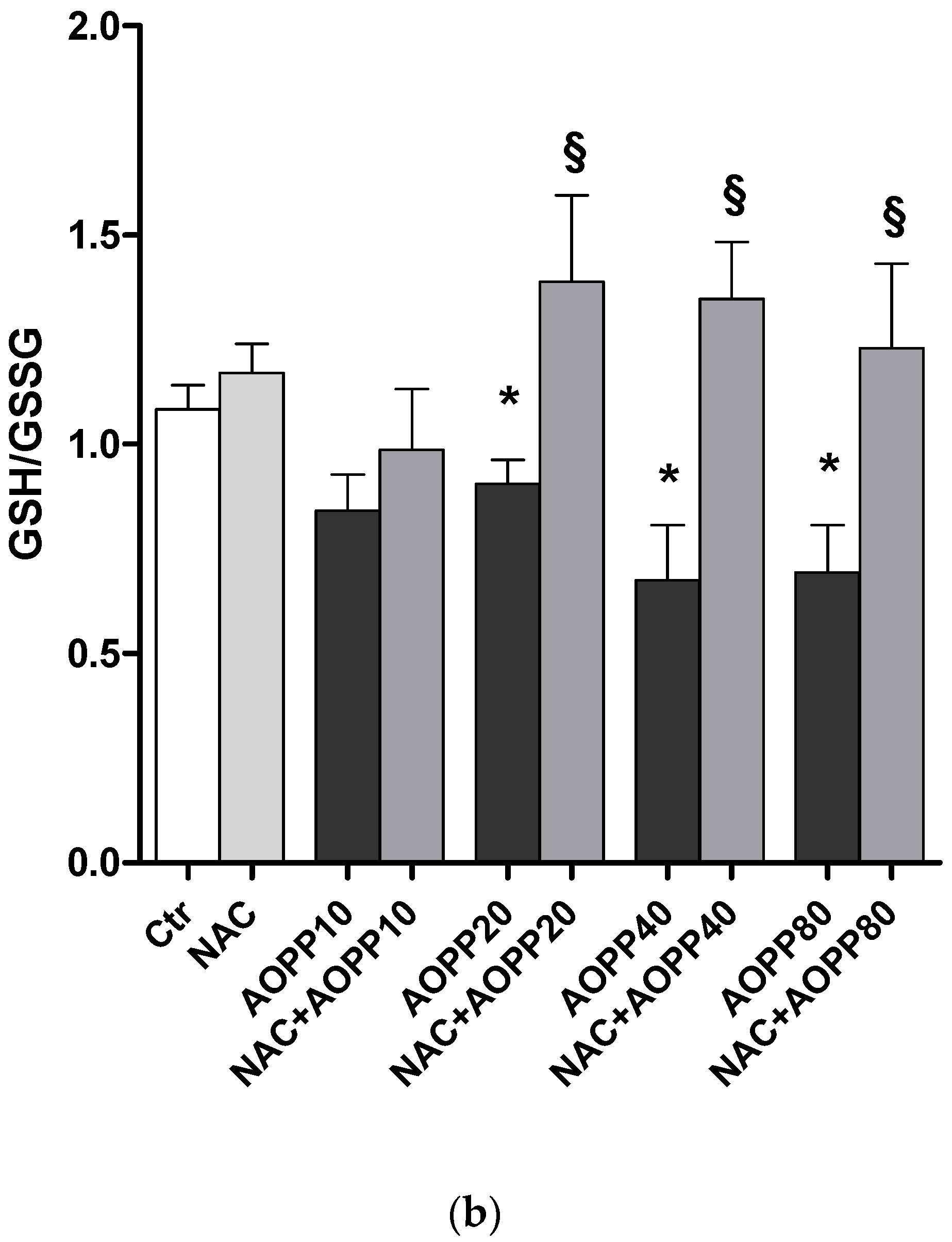
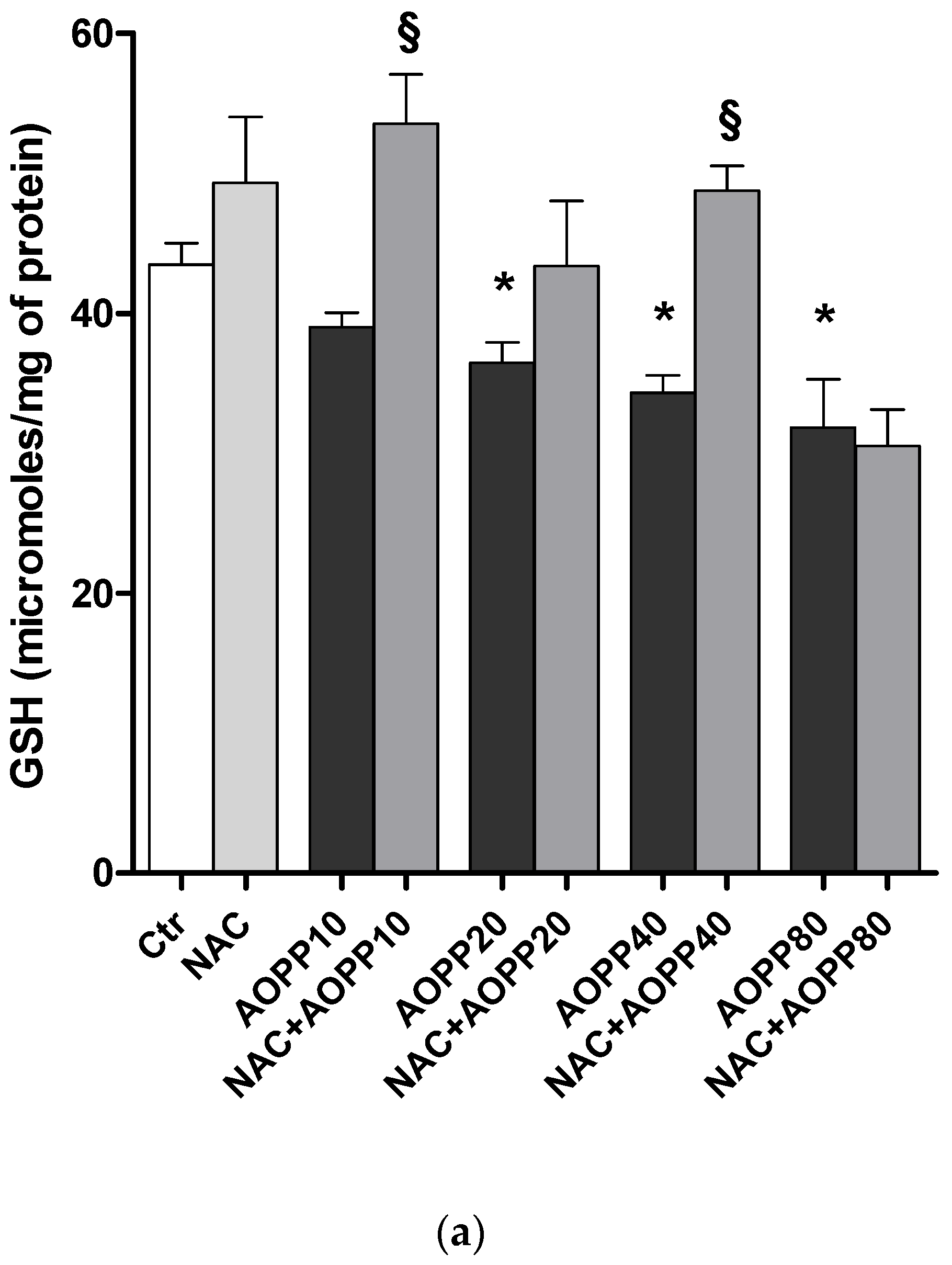
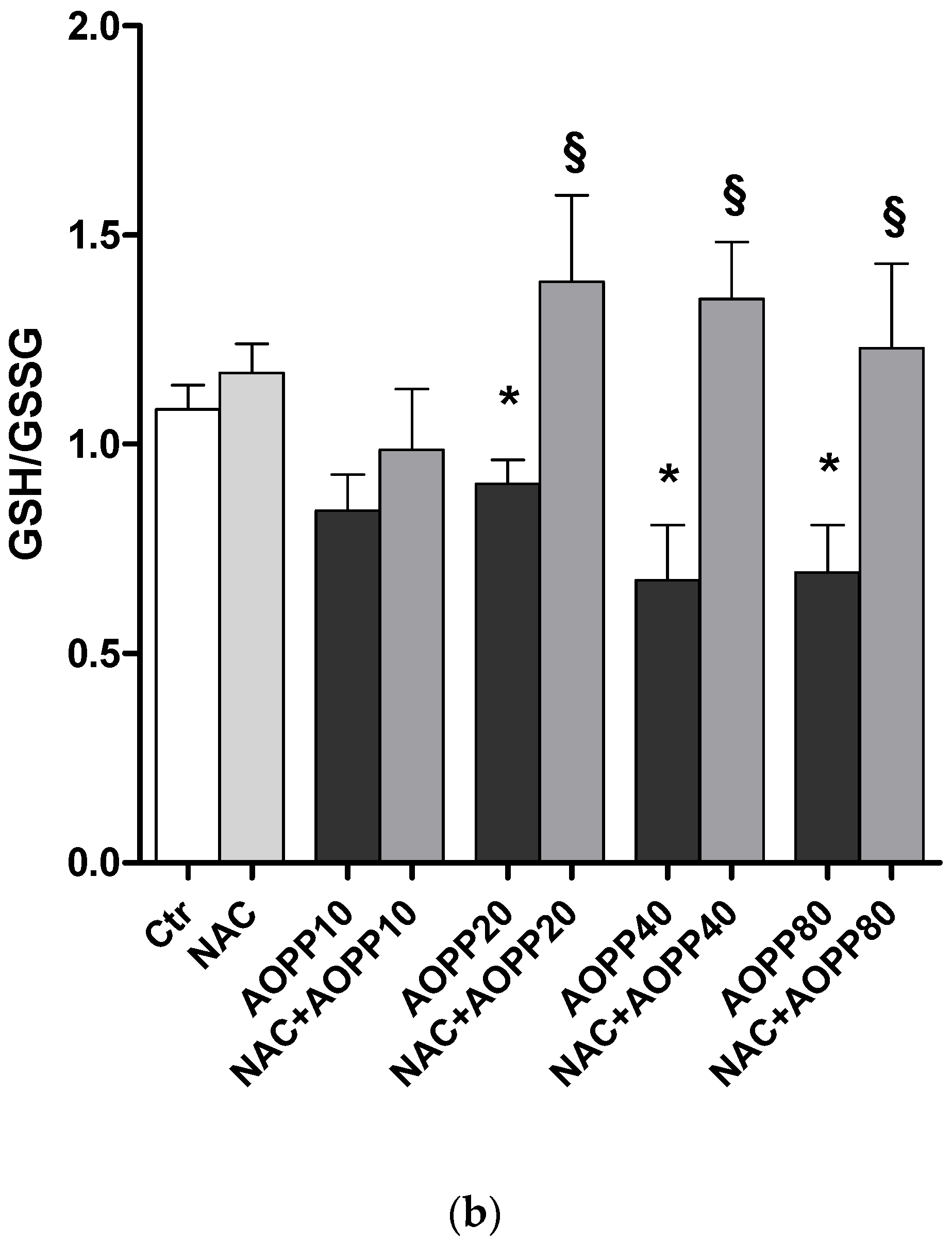
2.12. GSH and GSH/GSSG Ratio Decreased Following Treatment by HSA-AOPP in RAW264.7 Cells
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Materials
5.2. AOPP Preparation
5.3. AOPP Evaluation
5.4. Cells and Treatments
5.5. Flow Cytometry
5.6. Western Blotting
5.7. Glutathione Evaluation
5.8. Mixed Leukocyte Reactions (MLRs)
5.9. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Witko-Sarsat, V.; Friedlander, M.; Capeillere-Blandin, C.; Nguyen-Khoa, T.; Nguyen, A.T.; Zingraff, J.; Jungers, P.; Descamps-Latscha, B. Advanced oxidation protein products as a novel marker of oxidative stress in uremia. Kidney Int. 1996, 49, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Capeillere-Blandin, C.; Gausson, V.; Descamps-Latscha, B.; Witko-Sarsat, V. Biochemical and spectrophotometric significance of advanced oxidized protein products. Biochim. Biophys. Acta 2004, 1689, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Witko-Sarsat, V.; Descamps-Latscha, B. Advanced oxidation protein products: Novel uraemic toxins and pro-inflammatory mediators in chronic renal failure? Nephrol. Dial. Transplant. 1997, 12, 1310–1312. [Google Scholar] [CrossRef] [PubMed]
- Witko-Sarsat, V.; Gausson, V.; Descamps-Latscha, B. Are advanced oxidation protein products potential uremic toxins? Kidney Int. 2003, 84, S11–S14. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, H.; Taguchi, J.; Ogasawara, K.; Aizawa, T.; Ohno, M. Increased level of advanced oxidation protein products in patients with coronary artery disease. Atherosclerosis 2002, 162, 221–225. [Google Scholar] [CrossRef]
- Cakatay, U. Protein oxidation parameters in type 2 diabetic patients with good and poor glycaemic control. Diabetes Metab. 2005, 31, 551–557. [Google Scholar] [CrossRef]
- Shi, X.Y.; Hou, F.F.; Niu, H.X.; Wang, G.B.; Xie, D.; Guo, Z.J.; Zhou, Z.M.; Yang, F.; Tian, J.W.; Zhang, X. Advanced oxidation protein products promote inflammation in diabetic kidney through activation of renal nicotinamide adenine dinucleotide phosphate oxidase. Endocrinology 2008, 149, 1829–1839. [Google Scholar] [CrossRef] [PubMed]
- Servettaz, A.; Guilpain, P.; Goulvestre, C.; Chéreau, C.; Hercend, C.; Nicco, C.; Guillevin, L.; Weill, B.; Mouthon, L.; Batteux, F. Radical oxygen species production induced by advanced oxidation protein products predicts clinical evolution and response to treatment in systemic sclerosis. Ann. Rheum. Dis. 2007, 66, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Descamps-Latscha, B.; Witko-Sarsat, V.; Nguyen-Khoa, T.; Nguyen, A.T.; Gausson, V.; Mothu, N.; London, G.M.; Jungers, P. Advanced oxidation protein products as risk factors for atherosclerotic cardiovascular events in nondiabetic predialysis patients. Am. J. Kidney Dis. 2005, 45, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Yoshida, M. Protein-bound uremic toxins: New culprits of cardiovascular events in chronic kidney disease patients. Toxins 2014, 6, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Witko-Sarsat, V.; Gausson, V.; Nguyen, A.T.; Touam, M.; Drüeke, T.; Santangelo, F.; Descamps-Latscha, B. AOPP-induced activation of human neutrophil and monocyte oxidative metabolism: A potential target for N-acetylcysteine treatment in dialysis patients. Kidney Int. 2003, 64, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Witko-Sarsat, V.; Friedlander, M.; Capeillère-Blandin, C.; Nguyen-Khoa, T.; Nguyen, A.T.; Zingraff, J.; Jungers, P.; Descamps-Latscha, B. Advanced oxidation protein products as novel mediators of inflammation and monocyte activation in chronic renal failure. J. Immunol. 1998, 161, 2524–2532. [Google Scholar] [PubMed]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The dendritic cell lineage: Ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [PubMed]
- Hespel, C.; Moser, M. Role of inflammatory dendritic cells in innate and adaptive immunity. Eur. J. Immunol. 2012, 42, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- Dopheide, J.F.; Zeller, G.C.; Kuhlmann, M.; Girndt, M.; Sester, M.; Sester, U. Differentiation of Monocyte Derived Dendritic Cells in End Stage Renal Disease is Skewed towards Accelerated Maturation. Adv. Clin. Exp. Med. 2015, 24, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Heymann, F.; Meyer-Schwesinger, C.; Hamilton-Williams, E.E.; Hammerich, L.; Panzer, U.; Kaden, S.; Quaggin, S.E.; Floege, J.; Gröne, H.J.; Kurts, C. Kidney dendritic cell activation is required for progression of renal disease in a mouse model of glomerular injury. J. Clin. Investig. 2009, 119, 1286–1297. [Google Scholar] [CrossRef] [PubMed]
- Kitching, A.R. Dendritic cells in progressive renal disease: Some answers, many questions. Nephrol. Dial. Transplant. 2014, 29, 2185–2193. [Google Scholar] [CrossRef] [PubMed]
- Turell, L.; Radi, R.; Alvarez, B. The thiol pool in human plasma: The central contribution of albumin to redox processes. Free Radic. Biol. Med. 2013, 65, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Steinman, R.M. Dendritic cells: Specialized and regulated antigen processing machines. Cell 2001, 106, 255–258. [Google Scholar] [CrossRef]
- Seimon, T.A.; Nadolski, M.J.; Liao, X.; Magallon, J.; Nguyen, M.; Feric, N.T.; Koschinsky, M.L.; Harkewicz, R.; Witztum, J.L.; Tsimikas, S.; et al. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 2010, 12, 467–482. [Google Scholar] [CrossRef] [PubMed]
- Himmelfarb, J.; McMonagle, E. Albumin is the major plasma protein target of oxidant stress in uremia. Kidney Int. 2001, 60, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Anraku, M.; Chuang, V.T.; Maruyama, T.; Otagiri, M. Redox properties of serum albumin. Biochim. Biophys. Acta 2013, 1830, 5465–5472. [Google Scholar] [CrossRef] [PubMed]
- Pattison, D.I.; Hawkins, C.L.; Davies, M.J. What Are the Plasma Targets of the Oxidant Hypochlorous Acid? A Kinetic Modeling Approach. Chem. Res. Toxicol. 2009, 22, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Barsotti, A.; Fabbi, P.; Fedele, M.; Garibaldi, S.; Balbi, M.; Bezante, G.P.; Risso, D.; Indiveri, F.; Ghigliotti, G.; Brunelli, C. Role of advanced oxidation protein products and Thiol ratio in patients with acute coronary syndromes. Clin. Biochem. 2011, 44, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Spallarossa, P.; Garibaldi, S.; Barisione, C.; Ghigliotti, G.; Altieri, P.; Tracchi, I.; Fabbi, P.; Barsotti, A.; Brunelli, C. Postprandial serum induces apoptosis in endothelial cells: Role of polymorphonuclear-derived myeloperoxidase and metalloproteinase-9 activity. Atherosclerosis 2008, 198, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Biedroń, R.; Konopiński, M.K.; Marcinkiewicz, J.; Józefowski, S. Oxidation by neutrophils-derived HOCl increases immunogenicity of proteins by converting them into ligands of several endocytic receptors involved in antigen uptake by dendritic cells and macrophages. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.J.; Rees, A.J.; Griffin, M.D.; Hughes, J.; Kurts, C.; Duffield, J. The renal mononuclear phagocytic system. J. Am. Soc. Nephrol. 2012, 23, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Teteris, S.A.; Engel, D.R.; Kurts, C. Homeostatic and pathogenic role of renal dendritic cells. Kidney Int. 2011, 80, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Ozmen, J.; Bobryshev, Y.V.; Lord, R.S. CD40 co-stimulatory molecule expression by dendritic cells in primary atherosclerotic lesions in carotid arteries and in stenotic saphenous vein coronary artery grafts. Cardiovasc. Surg. 2001, 9, 329–333. [Google Scholar] [CrossRef]
- Sharma, R.; Li, D.; Zeng, Q.; Feng, Y.; Li, Y.; Wang, X.; Chao, L.S.; Tian, Y. Differentiation of dendritic cells in monocyte cultures isolated from patients with unstable angina. Int. J. Cardiol. 2004, 97, 551–555. [Google Scholar]
- Alderman, C.J.; Shah, S.; Foreman, J.C.; Chain, B.M.; Katz, D.R. The role of advanced oxidation protein products in regulation of dendritic cell function. Free Radic. Biol. Med. 2002, 32, 377–385. [Google Scholar] [CrossRef]
- Sahaf, B.; Heydari, K.; Herzenberg, L.A.; Herzenberg, L.A. Lymphocyte surface thiol levels. Proc. Natl. Acad. Sci. USA 2003, 100, 4001–4005. [Google Scholar] [CrossRef] [PubMed]
- Hirota, M.; Motoyama, A.; Suzuki, M.; Yanagi, M.; Kitagaki, M.; Kouzuki, H.; Hagino, S.; Itagaki, H.; Sasa, H.; Kagatani, S.; et al. Changes of cell-surface thiols and intracellular signaling in human monocytic cell line THP-1 treated with diphenylcyclopropenone. J. Toxicol. Sci. 2010, 35, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; Rotilio, G.; Ciriolo, M.R. Glutathione disulfide induces apoptosis in U937 cells by a redox-mediated p38 MAP kinase pathway. FASEB J. 2003, 17, 64–66. [Google Scholar] [PubMed]
- Tanaka, T.; Nakamura, H.; Yodoi, J.; Bloom, E.T. Redox regulation of the signaling pathways leading to eNOS phosphorylation. Free. Radic. Biol. Med. 2005, 38, 1231–1242. [Google Scholar] [CrossRef] [PubMed]
- Ashfaq, S.; Abramson, J.L.; Jones, D.P.; Rhodes, S.D.; Weintraub, W.S.; Hooper, W.C.; Vaccarino, V.; Harrison, D.G.; Quyyumi, A.A. The relationship between plasma levels of oxidized and reduced thiols and early atherosclerosis in healthy adults. J. Am. Coll. Cardiol. 2006, 47, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.M.; Jones, D.P. Intracellular proatherogenic events and cell adhesion modulated by extracellular thiol/disulfide redox state. Circulation 2005, 111, 2973–2980. [Google Scholar] [CrossRef] [PubMed]
- Ghigliotti, G.; Mereto, E.; Eisenberg, P.R.; Martelli, A.; Orsi, P.; Sini, D.; Spallarossa, P.; Olivotti, L.; Brunelli, C. N-acetyl-cysteine reduces neointimal thickening and procoagulant activity after balloon-induced injury in abdominal aortae of New Zealand white rabbits. Thromb. Haemost. 2001, 85, 724–729. [Google Scholar] [PubMed]
- Demicheli, G.; Zanini, P.; Bertocchi, F.; Falcone, C.; Ghio, S.; Marinoni, G.; Montemartini, C.; Mussini, A. Effect of transdermal nitroglycerin or N-acetylcysteine, or both, in the long-term treatment of unstable angina pectoris. J. Am. Coll. Cardiol. 1997, 29, 941–947. [Google Scholar]
- Cao, W.; Xu, J.; Zhou, Z.M.; Wang, G.B.; Hou, F.F.; Nie, J. Advanced oxidation protein products activate intrarenal renin-angiotensin system via a CD36-mediated, redox-dependent pathway. Antioxid. Redox Signal 2013, 18, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Hanasand, M.; Omdal, R.; Norheim, K.B.; Gøransson, L.G.; Brede, C.; Jonsson, G. Improved detection of advanced oxidation protein products in plasma. Clin. Chim. Acta 2012, 413, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Colla, R.; Izzotti, A.; De Ciucis, C.; Fenoglio, D.; Ravera, S.; Speciale, A.; Ricciarelli, R.; Furfaro, A.L.; Pulliero, A.; Passalacqua, M.; et al. Glutathione-mediated antioxidant response and aerobic metabolism: Two crucial factors involved in determining the multi-drug resistance of high-risk neuroblastoma. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garibaldi, S.; Barisione, C.; Marengo, B.; Ameri, P.; Brunelli, C.; Balbi, M.; Ghigliotti, G. Advanced Oxidation Protein Products-Modified Albumin Induces Differentiation of RAW264.7 Macrophages into Dendritic-Like Cells Which Is Modulated by Cell Surface Thiols. Toxins 2017, 9, 27. https://doi.org/10.3390/toxins9010027
Garibaldi S, Barisione C, Marengo B, Ameri P, Brunelli C, Balbi M, Ghigliotti G. Advanced Oxidation Protein Products-Modified Albumin Induces Differentiation of RAW264.7 Macrophages into Dendritic-Like Cells Which Is Modulated by Cell Surface Thiols. Toxins. 2017; 9(1):27. https://doi.org/10.3390/toxins9010027
Chicago/Turabian StyleGaribaldi, Silvano, Chiara Barisione, Barbara Marengo, Pietro Ameri, Claudio Brunelli, Manrico Balbi, and Giorgio Ghigliotti. 2017. "Advanced Oxidation Protein Products-Modified Albumin Induces Differentiation of RAW264.7 Macrophages into Dendritic-Like Cells Which Is Modulated by Cell Surface Thiols" Toxins 9, no. 1: 27. https://doi.org/10.3390/toxins9010027
APA StyleGaribaldi, S., Barisione, C., Marengo, B., Ameri, P., Brunelli, C., Balbi, M., & Ghigliotti, G. (2017). Advanced Oxidation Protein Products-Modified Albumin Induces Differentiation of RAW264.7 Macrophages into Dendritic-Like Cells Which Is Modulated by Cell Surface Thiols. Toxins, 9(1), 27. https://doi.org/10.3390/toxins9010027