
Shiga Toxin Therapeutics: Beyond Neutralization

Abstract
:1. Introduction
2. Shiga Toxin Structure and Activity
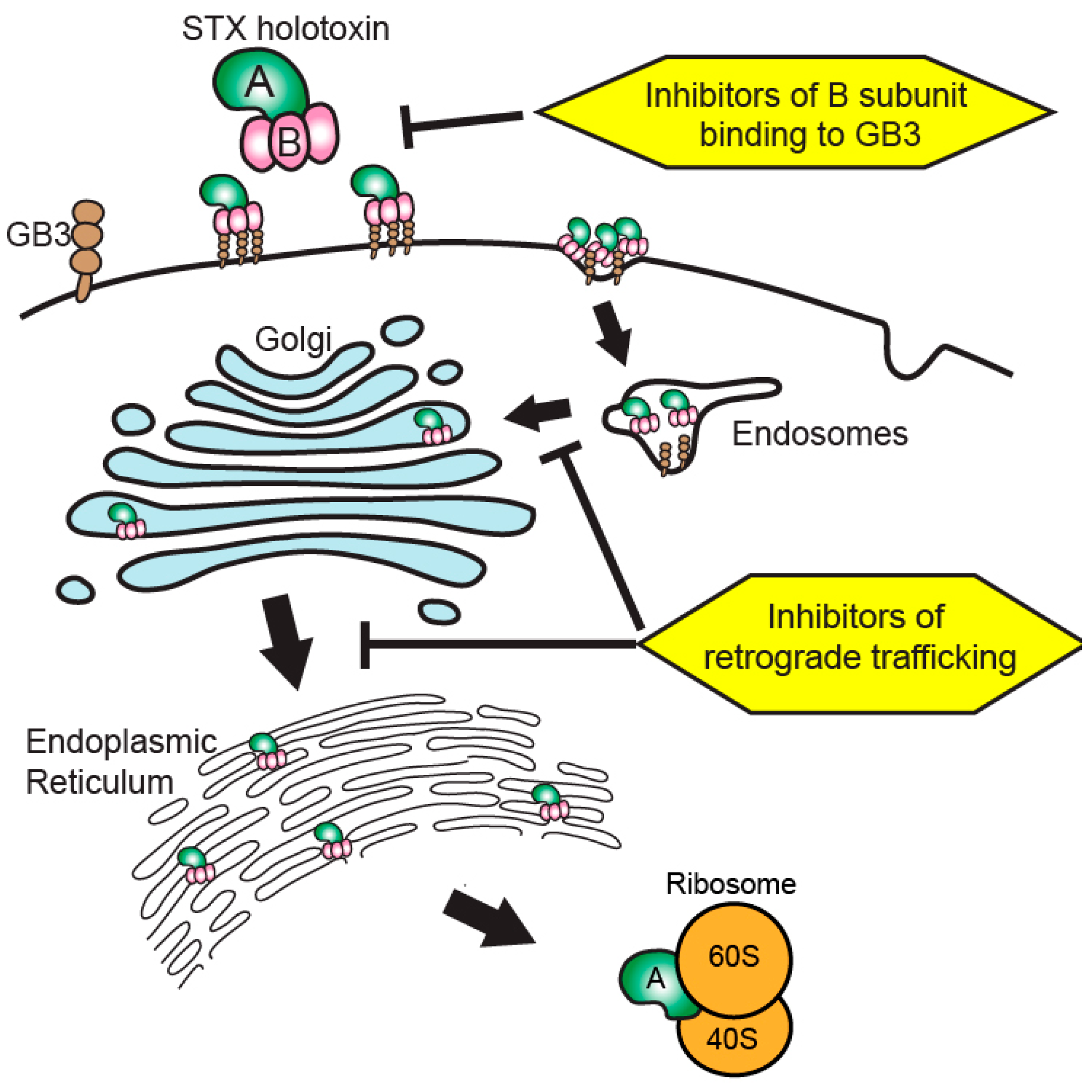
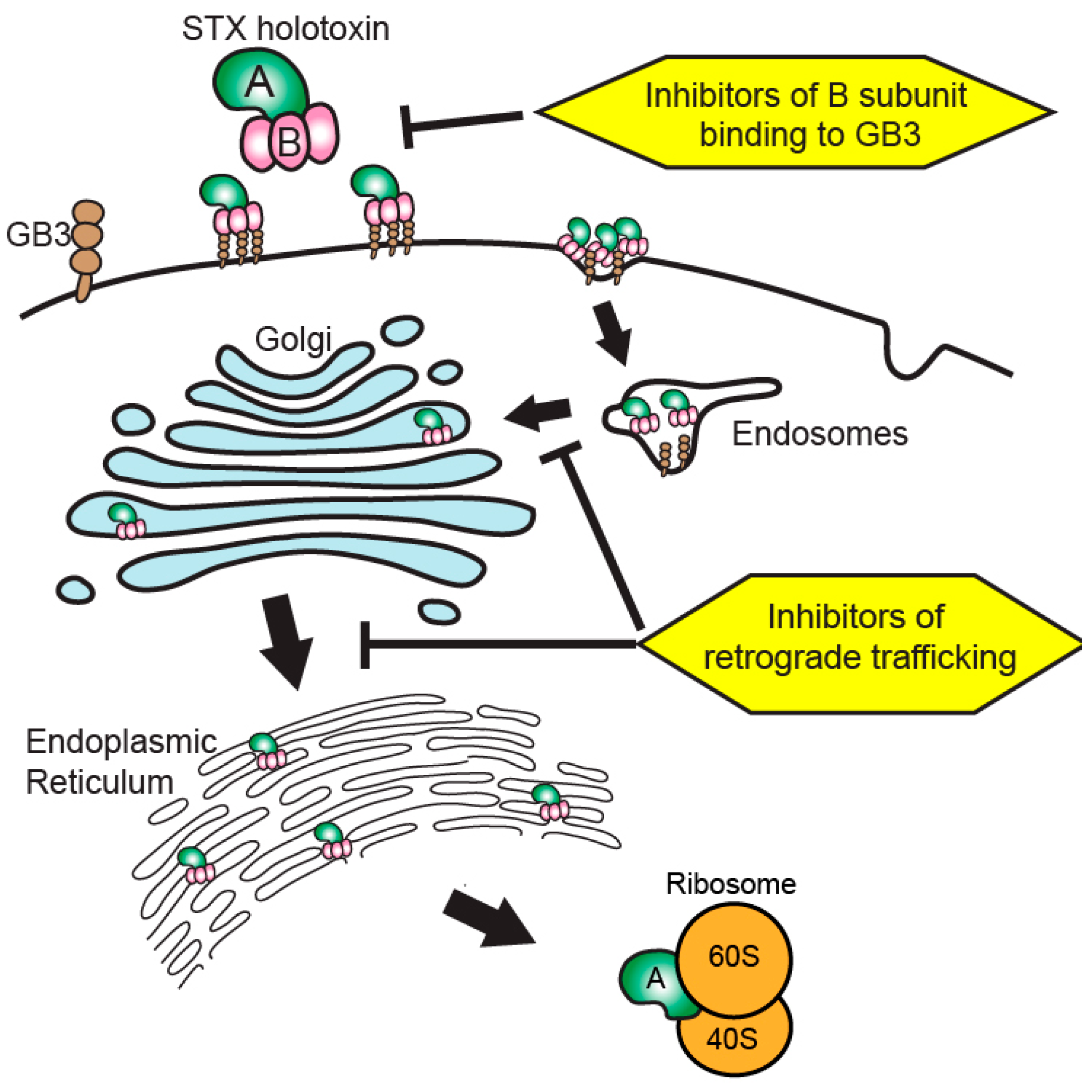
3. Current State of Shiga Toxin-Targeted Therapeutic Development
4. Beyond Toxin Neutralization
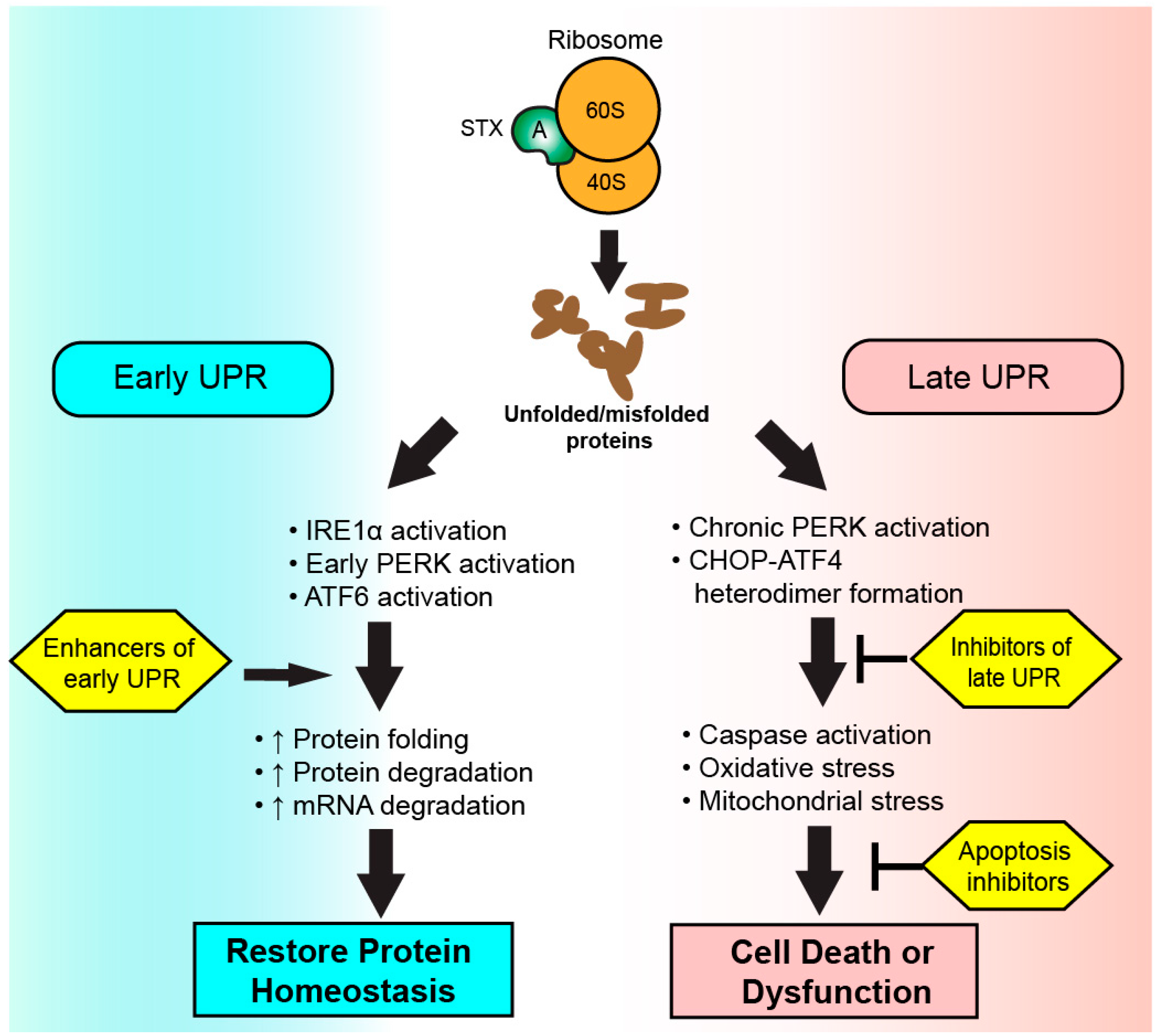
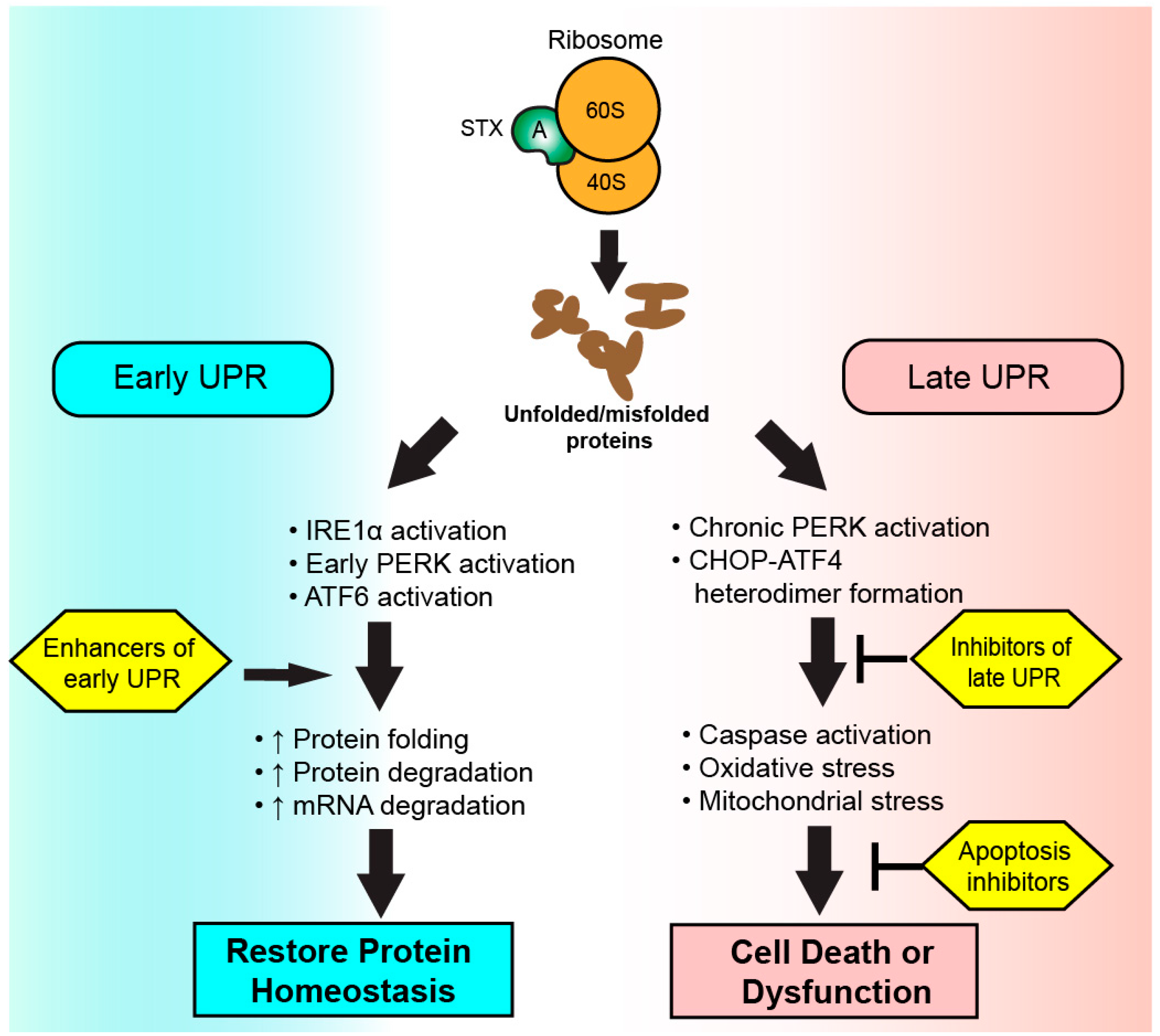
5. The Unfolded Protein Response (UPR)
6. The UPR in Health, Disease, and Shiga Toxicosis
7. Targeting the UPR
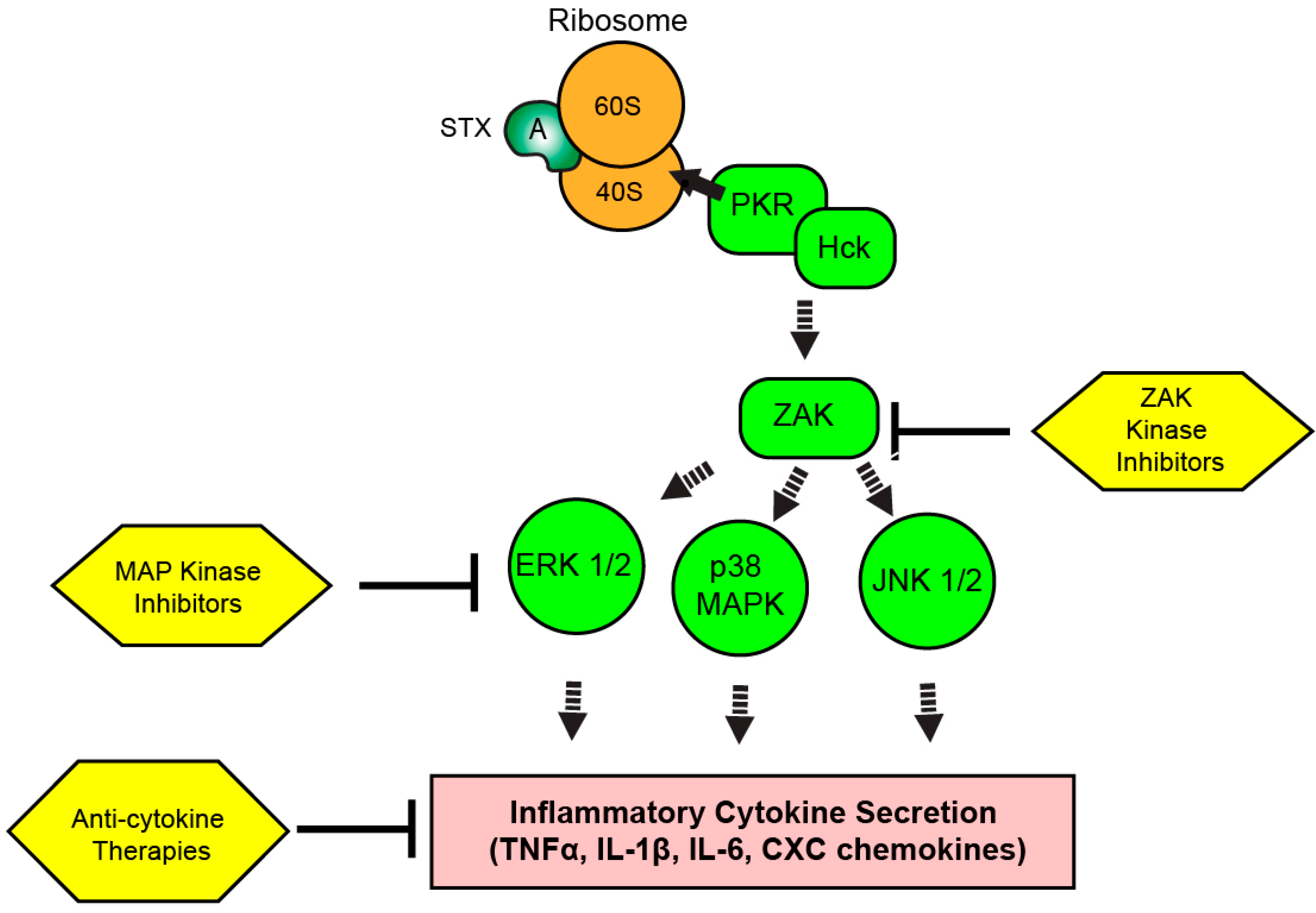
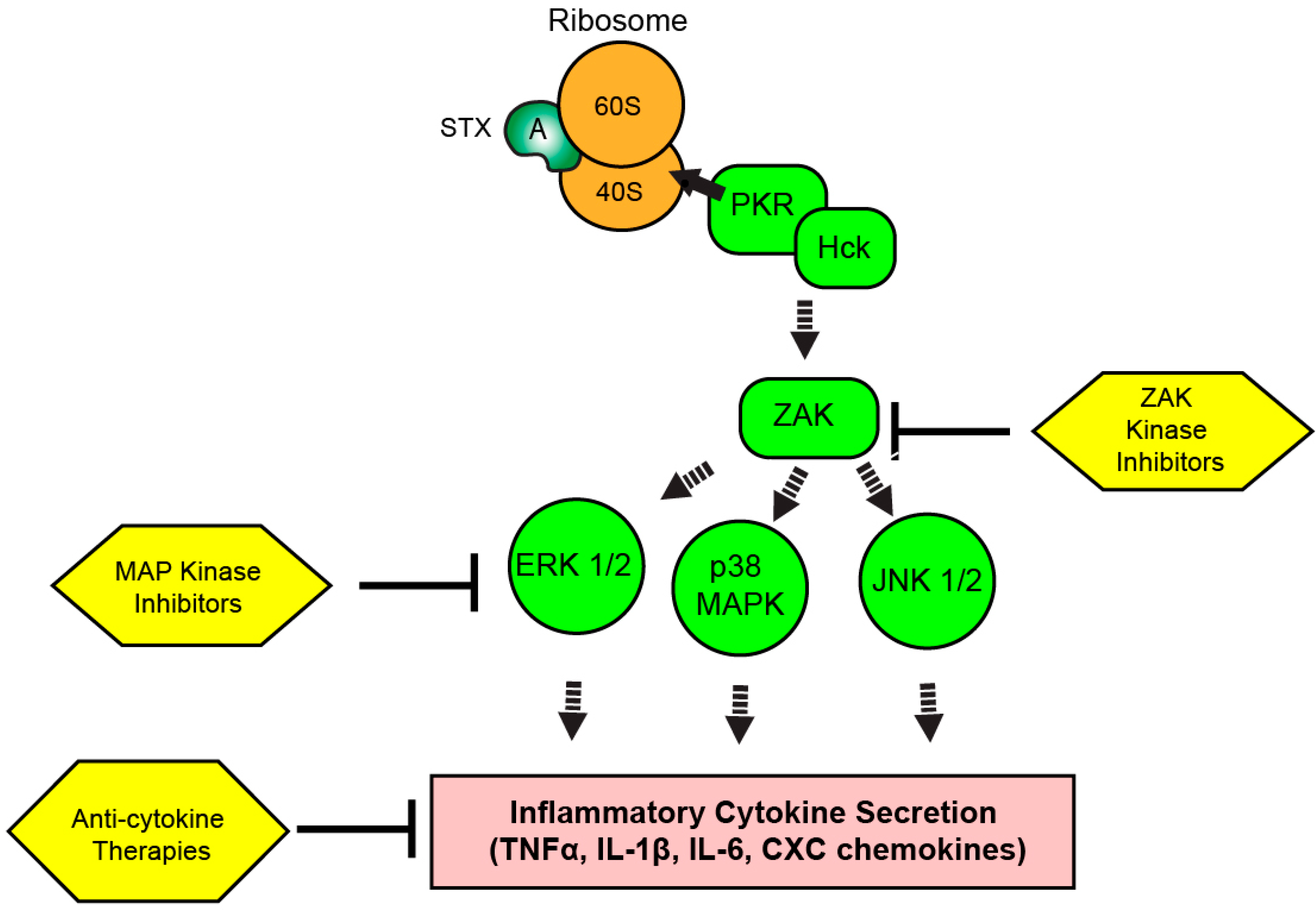
8. The Ribotoxic Response
9. Targeting the RSR and Inflammation during Shiga Toxicosis
10. Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Krüger, A.; Lucchesi, P.M.A. Shiga toxins and stx phages: highly diverse entities. Microbiology 2015, 161, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Melton-Celsa, A.R. Shiga Toxin (Stx) Classification, Structure, and Function. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Njamkepo, E.; Fawal, N.; Tran-Dien, A.; Hawkey, J.; Strockbine, N.; Jenkins, C.; Talukder, K.A.; Bercion, R.; Kuleshov, K.; Kolínská, R.; et al. Global phylogeography and evolutionary history of Shigella dysenteriae type 1. Nat. Microbiol. 2016, 1, 16027. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M.B.; Estrada-García, T. Shigella: A Highly Virulent and Elusive Pathogen. Curr. Trop. Med. Rep. 2014, 1, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Rangel, J.M.; Sparling, P.H.; Crowe, C.; Griffin, P.M.; Swerdlow, D.L. Epidemiology of Escherichia coli O157:H7 Outbreaks, United States, 1982–2002. Emerg. Infect. Dis. 2005, 11, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Frank, C.; Werber, D.; Cramer, J.P.; Askar, M.; Faber, M.; an der Heiden, M.; Bernard, H.; Fruth, A.; Prager, R.; Spode, A.; et al. Epidemic Profile of Shiga-Toxin–Producing Escherichia coli O104:H4 Outbreak in Germany. N. Engl. J. Med. 2011, 365, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Terajima, J.; Iyoda, S.; Ohnishi, M.; Watanabe, H. Shiga Toxin (Verotoxin)-Producing Escherichia coli in Japan. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, A.D.; Newland, J.W.; Miller, S.F.; Holmes, R.K.; Smith, H.W.; Formal, S.B. Shiga-like toxin-converting phages from Escherichia coli strains that cause hemorrhagic colitis or infantile diarrhea. Science 1984, 226, 694–696. [Google Scholar] [CrossRef] [PubMed]
- Karmali, M.A.; Steele, B.T.; Petric, M.; Lim, C. Sporadic cases of haemolytic-uraemic syndrome associated with faecal cytotoxin and cytotoxin-producing Escherichia coli in stools. Lancet Lond. Engl. 1983, 1, 619–620. [Google Scholar] [CrossRef]
- Mayer, C.L.; Leibowitz, C.S.; Kurosawa, S.; Stearns-Kurosawa, D.J. Shiga toxins and the pathophysiology of hemolytic uremic syndrome in humans and animals. Toxins 2012, 4, 1261–1287. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.L.F.; Applegate, J.A.; Black, R.E. Haemolytic-Uraemic Syndrome as a Sequela of Diarrhoeal Disease. J. Health Popul. Nutr. 2012, 30, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Karpman, D.; Loos, S.; Tati, R.; Arvidsson, I. Haemolytic uraemic syndrome. J. Intern. Med. 2017, 281, 123–148. [Google Scholar] [CrossRef] [PubMed]
- Weissenborn, K.; Donnerstag, F.; Kielstein, J.T.; Heeren, M.; Worthmann, H.; Hecker, H.; Schmitt, R.; Schiffer, M.; Pasedag, T.; Schuppner, R.; et al. Neurologic manifestations of E coli infection-induced hemolytic-uremic syndrome in adults. Neurology 2012, 79, 1466–1473. [Google Scholar] [CrossRef] [PubMed]
- Takanashi, J.; Taneichi, H.; Misaki, T.; Yahata, Y.; Okumura, A.; Ishida, Y.; Miyawaki, T.; Okabe, N.; Sata, T.; Mizuguchi, M. Clinical and radiologic features of encephalopathy during 2011 E coli O111 outbreak in Japan. Neurology 2014, 82, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Rosales, A.; Hofer, J.; Zimmerhackl, L.-B.; Jungraithmayr, T.C.; Riedl, M.; Giner, T.; Strasak, A.; Orth-Höller, D.; Würzner, R.; Karch, H.; et al. Need for Long-term Follow-up in Enterohemorrhagic Escherichia coli–Associated Hemolytic Uremic Syndrome Due to Late-Emerging Sequelae. Clin. Infect. Dis. 2012, 54, 1413–1421. [Google Scholar] [CrossRef] [PubMed]
- Scallan, E.; Mahon, B.E.; Hoekstra, R.M.; Griffin, P.M. Estimates of Illnesses, Hospitalizations, and Deaths Caused By Major Bacterial Enteric Pathogens in Young Children in the United States. Pediatr. Infect. Dis. J. 2012, 32, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Bielaszewska, M.; Mellmann, A.; Zhang, W.; Köck, R.; Fruth, A.; Bauwens, A.; Peters, G.; Karch, H. Characterisation of the Escherichia coli strain associated with an outbreak of haemolytic uraemic syndrome in Germany, 2011: a microbiological study. Lancet Infect. Dis. 2011, 11, 671–676. [Google Scholar] [CrossRef]
- Wong, C.S.; Jelacic, S.; Habeeb, R.L.; Watkins, S.L.; Tarr, P.I. The risk of the hemolytic–uremic syndrome after antibiotic treatment of escherichia coli o157:h7 infections. N. Engl. J. Med. 2000, 342, 1930–1936. [Google Scholar] [CrossRef] [PubMed]
- Freedman, S.B.; Xie, J.; Neufeld, M.S.; Hamilton, W.L.; Hartling, L.; Tarr, P.I.; Nettel-Aguirre, A.; Chuck, A.; Lee, B.; Johnson, D.; et al. Shiga Toxin–Producing Escherichia coli Infection, Antibiotics, and Risk of Developing Hemolytic Uremic Syndrome: A Meta-analysis. Clin. Infect. Dis. 2016, 62, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Corogeanu, D.; Willmes, R.; Wolke, M.; Plum, G.; Utermöhlen, O.; Krönke, M. Therapeutic concentrations of antibiotics inhibit Shiga toxin release from enterohemorrhagic E. coli O104:H4 from the 2011 German outbreak. BMC Microbiol. 2012, 12, 160. [Google Scholar] [CrossRef] [PubMed]
- Ardissino, G.; Tel, F.; Possenti, I.; Testa, S.; Consonni, D.; Paglialonga, F.; Salardi, S.; Borsa-Ghiringhelli, N.; Salice, P.; Tedeschi, S.; et al. Early Volume Expansion and Outcomes of Hemolytic Uremic Syndrome. Pediatrics 2016, 137, e20152153. [Google Scholar] [CrossRef] [PubMed]
- Delmas, Y.; Vendrely, B.; Clouzeau, B.; Bachir, H.; Bui, H.-N.; Lacraz, A.; Hélou, S.; Bordes, C.; Reffet, A.; Llanas, B.; et al. Outbreak of Escherichia coli O104:H4 haemolytic uraemic syndrome in France: Outcome with eculizumab. Nephrol. Dial. Transplant. 2014, 29, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Kielstein, J.T.; Beutel, G.; Fleig, S.; Steinhoff, J.; Meyer, T.N.; Hafer, C.; Kuhlmann, U.; Bramstedt, J.; Panzer, U.; Vischedyk, M.; et al. Collaborators of the DGfN STEC-HUS registry Best supportive care and therapeutic plasma exchange with or without eculizumab in Shiga-toxin-producing E. coli O104:H4 induced haemolytic-uraemic syndrome: An analysis of the German STEC-HUS registry. Nephrol. Dial. Transplant. 2012, 27, 3807–3815. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, A.D.; Melton-Celsa, A.R. New Therapeutic Developments against Shiga Toxin-Producing Escherichia coli. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef]
- Fraser, M.E.; Fujinaga, M.; Cherney, M.M.; Melton-Celsa, A.R.; Twiddy, E.M.; O’Brien, A.D.; James, M.N.G. Structure of Shiga Toxin Type 2 (Stx2) from Escherichia coli O157:H7. J. Biol. Chem. 2004, 279, 27511–27517. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Boodhoo, A.; Hazes, B.; Cummings, M.D.; Armstrong, G.D.; Brunton, J.L.; Read, R.J. Structure of the Shiga-like Toxin I B-Pentamer Complexed with an Analogue of Its Receptor Gb3. Biochemistry (Mosc.) 1998, 37, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Lauvrak, S.U.; Torgersen, M.L.; Sandvig, K. Efficient endosome-to-Golgi transport of Shiga toxin is dependent on dynamin and clathrin. J. Cell Sci. 2004, 117, 2321–2331. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Bergan, J.; Dyve, A.-B.; Skotland, T.; Torgersen, M.L. Endocytosis and retrograde transport of Shiga toxin. Toxicon 2010, 56, 1181–1185. [Google Scholar] [CrossRef] [PubMed]
- Garred, O.; van Deurs, B.; Sandvig, K. Furin-induced Cleavage and Activation of Shiga Toxin. J. Biol. Chem. 1995, 270, 10817–10821. [Google Scholar] [CrossRef] [PubMed]
- Garred, Ø.; Dubinina, E.; Holm, P.K.; Olsnes, S.; Van Deurs, B.; Kozlov, J.V.; Sandvig, K. Role of Processing and Intracellular Transport for Optimal Toxicity of Shiga Toxin and Toxin Mutants. Exp. Cell Res. 1995, 218, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Tsurugi, K.; Yutsudo, T.; Takeda, Y.; Ogasawara, T.; Igarashi, K. Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. Eur. J. Biochem. 1988, 171, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.-W.; Mak, A.N.-S.; Wong, K.-B.; Shaw, P.-C. Structures and Ribosomal Interaction of Ribosome-Inactivating Proteins. Molecules 2016, 21, 1588. [Google Scholar] [CrossRef] [PubMed]
- Furutani, M.; Kashiwagi, K.; Ito, K.; Endo, Y.; Igarashi, K. Comparison of the modes of action of a vero toxin (a Shiga-like toxin) from Escherichia coli, of ricin, and of α-sarcin. Arch. Biochem. Biophys. 1992, 293, 140–146. [Google Scholar] [CrossRef]
- Lentz, E.K.; Leyva-Illades, D.; Lee, M.-S.; Cherla, R.P.; Tesh, V.L. Differential Response of the Human Renal Proximal Tubular Epithelial Cell Line HK-2 to Shiga Toxin Types 1 and 2. Infect. Immun. 2011, 79, 3527–3540. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; Cherla, R.P.; Caliskan, I.; Tesh, V.L. Shiga Toxin 1 Induces Apoptosis in the Human Myelogenous Leukemia Cell Line THP-1 by a Caspase-8-Dependent, Tumor Necrosis Factor Receptor-Independent Mechanism. Infect. Immun. 2005, 73, 5115–5126. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, S.W.; Samuel, J.E.; Schmitt, C.K.; O’Brien, A.D. The specific activities of Shiga-like toxin type II (SLT-II) and SLT-II-related toxins of enterohemorrhagic Escherichia coli differ when measured by Vero cell cytotoxicity but not by mouse lethality. Infect. Immun. 1994, 62, 623–631. [Google Scholar] [PubMed]
- García, A.; Bosques, C.J.; Wishnok, J.S.; Feng, Y.; Karalius, B.J.; Butterton, J.R.; Schauer, D.B.; Rogers, A.B.; Fox, J.G. Renal Injury Is a Consistent Finding in Dutch Belted Rabbits Experimentally Infected with Enterohemorrhagic Escherichia coli. J. Infect. Dis. 2006, 193, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.E.; Rotman, T.A.; Jay, V.; Smith, C.R.; Becker, L.E.; Petric, M.; Olivieri, N.F.; Karmali, M.A. Experimental verocytotoxemia in rabbits. Infect. Immun. 1992, 60, 4154–4167. [Google Scholar] [PubMed]
- Tesh, V.L.; Burris, J.A.; Owens, J.W.; Gordon, V.M.; Wadolkowski, E.A.; O’Brien, A.D.; Samuel, J.E. Comparison of the relative toxicities of Shiga-like toxins type I and type II for mice. Infect. Immun. 1993, 61, 3392–3402. [Google Scholar] [PubMed]
- Stearns-Kurosawa, D.J.; Oh, S.-Y.; Cherla, R.P.; Lee, M.-S.; Tesh, V.L.; Papin, J.; Henderson, J.; Kurosawa, S. Distinct Renal Pathology and a Chemotactic Phenotype after Enterohemorrhagic Escherichia coli Shiga Toxins in Non-Human Primate Models of Hemolytic Uremic Syndrome. Am. J. Pathol. 2013, 182, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-S.; Kwon, H.; Lee, E.-Y.; Kim, D.-J.; Park, J.-H.; Tesh, V.L.; Oh, T.-K.; Kim, M.H. Shiga Toxins Activate the NLRP3 Inflammasome Pathway To Promote Both Production of the Proinflammatory Cytokine Interleukin-1β and Apoptotic Cell Death. Infect. Immun. 2016, 84, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.M.; van Haaften, W.C.E.; Tesh, V.L. Regulation of Proinflammatory Cytokine Expression by Shiga Toxin 1 and/or Lipopolysaccharides in the Human Monocytic Cell Line THP-1. Infect. Immun. 2004, 72, 2618–2627. [Google Scholar] [CrossRef] [PubMed]
- Leyva-Illades, D.; Cherla, R.P.; Lee, M.-S.; Tesh, V.L. Regulation of Cytokine and Chemokine Expression by the Ribotoxic Stress Response Elicited by Shiga Toxin Type 1 in Human Macrophage-Like THP-1 Cells. Infect. Immun. 2012, 80, 2109–2120. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; Lee, M.-S.; Cherla, R.P.; Tesh, V.L. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell. Microbiol. 2008, 10, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, D.M.; Ahluwalia, A.; Schimmel, J.J.; Rogers, A.B.; Leong, J.M.; Thorpe, C.M. Activation of the Classical Mitogen-Activated Protein Kinases Is Part of the Shiga Toxin-Induced Ribotoxic Stress Response and May Contribute to Shiga Toxin-Induced Inflammation. Infect. Immun. 2016, 84, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Parello, C.; Mayer, C.; Lee, B.; Motomochi, A.; Kurosawa, S.; Stearns-Kurosawa, D. Shiga Toxin 2-Induced Endoplasmic Reticulum Stress Is Minimized by Activated Protein C but Does Not Correlate with Lethal Kidney Injury. Toxins 2015, 7, 170–186. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.; Pillai, S.K.; Bower, W.A.; Hendricks, K.A.; Guarnizo, J.T.; Hoyle, J.D.; Gorman, S.E.; Boyer, A.E.; Quinn, C.P.; Meaney-Delman, D. Antitoxin Treatment of Inhalation Anthrax: A Systematic Review. Health Secur. 2015, 13, 365. [Google Scholar] [CrossRef] [PubMed]
- Lukić, I.; Marinković, E.; Filipović, A.; Krnjaja, O.; Kosanović, D.; Inić-Kanada, A.; Stojanović, M. Key protection factors against tetanus: Anti-tetanus toxin antibody affinity and its ability to prevent tetanus toxin—Ganglioside interaction. Toxicon 2015, 103, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Donohue-Rolfe, A.; Kondova, I.; Mukherjee, J.; Chios, K.; Hutto, D.; Tzipori, S. Antibody-Based Protection of Gnotobiotic Piglets Infected with Escherichia coli O157:H7 against Systemic Complications Associated with Shiga Toxin 2. Infect. Immun. 1999, 67, 3645–3648. [Google Scholar] [PubMed]
- Yamagami, S.; Motoki, M.; Kimura, T.; Izumi, H.; Takeda, T.; Katsuura, Y.; Matsumoto, Y. Efficacy of Postinfection Treatment with Anti-Shiga Toxin (Stx) 2 Humanized Monoclonal Antibody TMA-15 in Mice Lethally Challenged with Stx-Producing Escherichia coli. J. Infect. Dis. 2001, 184, 738–742. [Google Scholar] [CrossRef] [PubMed]
- Sheoran, A.S.; Chapman, S.; Singh, P.; Donohue-Rolfe, A.; Tzipori, S. Stx2-Specific Human Monoclonal Antibodies Protect Mice against Lethal Infection with Escherichia coli Expressing Stx2 Variants. Infect. Immun. 2003, 71, 3125–3130. [Google Scholar] [CrossRef] [PubMed]
- Sauter, K.A.D.; Melton-Celsa, A.R.; Larkin, K.; Troxell, M.L.; O’Brien, A.D.; Magun, B.E. Mouse Model of Hemolytic-Uremic Syndrome Caused by Endotoxin-Free Shiga Toxin 2 (Stx2) and Protection from Lethal Outcome by Anti-Stx2 Antibody. Infect. Immun. 2008, 76, 4469–4478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, E.L.; Contrini, M.M.; Glatstein, E.; Ayala, S.G.; Santoro, R.; Allende, D.; Ezcurra, G.; Teplitz, E.; Koyama, T.; Matsumoto, Y.; et al. Safety and Pharmacokinetics of Urtoxazumab, a Humanized Monoclonal Antibody, against Shiga-Like Toxin 2 in Healthy Adults and in Pediatric Patients Infected with Shiga-Like Toxin-Producing Escherichia coli. Antimicrob. Agents Chemother. 2010, 54, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Dowling, T.C.; Chavaillaz, P.A.; Young, D.G.; Melton-Celsa, A.; O’Brien, A.; Thuning-Roberson, C.; Edelman, R.; Tacket, C.O. Phase 1 Safety and Pharmacokinetic Study of Chimeric Murine-Human Monoclonal Antibody cαStx2 Administered Intravenously to Healthy Adult Volunteers. Antimicrob. Agents Chemother. 2005, 49, 1808–1812. [Google Scholar] [CrossRef] [PubMed]
- Mejías, M.P.; Hiriart, Y.; Lauché, C.; Fernández-Brando, R.J.; Pardo, R.; Bruballa, A.; Ramos, M.V.; Goldbaum, F.A.; Palermo, M.S.; Zylberman, V. Development of camelid single chain antibodies against Shiga toxin type 2 (Stx2) with therapeutic potential against Hemolytic Uremic Syndrome (HUS). Sci. Rep. 2016, 6, 24913. [Google Scholar] [CrossRef] [PubMed]
- Sheoran, A.S.; Dmitriev, I.P.; Kashentseva, E.A.; Cohen, O.; Mukherjee, J.; Debatis, M.; Shearer, J.; Tremblay, J.M.; Beamer, G.; Curiel, D.T.; et al. Adenovirus Vector Expressing Stx1/Stx2-Neutralizing Agent Protects Piglets Infected with Escherichia coli O157:H7 against Fatal Systemic Intoxication. Infect. Immun. 2015, 83, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, G.D.; Rowe, P.C.; Goodyer, P.; Orrbine, E.; Klassen, T.P.; Wells, G.; MacKenzie, A.; Lior, H.; Blanchard, C.; Auclair, F.; et al. A Phase I Study of Chemically Synthesized Verotoxin (Shiga-like Toxin) Pk-Trisaccharide Receptors Attached to Chromosorb for Preventing Hemolytic-Uremic Syndrome. J. Infect. Dis. 1995, 171, 1042–1045. [Google Scholar] [CrossRef] [PubMed]
- Trachtman, H.; Cnaan, A.; Christen, E.; Gibbs, K.; Zhao, S.; Acheson, D.W.K.; Weiss, R.; Kaskel, F.J.; Spitzer, A.; Hirschman, G.H. Investigators of the HUS-SYNSORB Pk Multicenter Clinical Trial Effect of an oral Shiga toxin-binding agent on diarrhea-associated hemolytic uremic syndrome in children: a randomized controlled trial. JAMA 2003, 290, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Watanabe-Takahashi, M.; Shimizu, E.; Nishikawa, K. Identification of a Wide Range of Motifs Inhibitory to Shiga Toxin by Affinity-Driven Screening of Customized Divalent Peptides Synthesized on a Membrane. Appl. Environ. Microbiol. 2015, 81, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, T.; Watanabe-Takahashi, M.; Shimizu, E.; Zhang, B.; Funamoto, S.; Yamasaki, S.; Nishikawa, K. Affinity-based screening of tetravalent peptides identifies subtype-selective neutralizers of Shiga toxin 2d, a highly virulent subtype, by targeting a unique amino acid involved in its receptor recognition. Infect. Immun. 2016, 84, 2653–2661, IAI.00149-16. [Google Scholar] [CrossRef] [PubMed]
- Stearns-Kurosawa, D.J.; Collins, V.; Freeman, S.; Debord, D.; Nishikawa, K.; Oh, S.-Y.; Leibowitz, C.S.; Kurosawa, S. Rescue from lethal Shiga toxin 2-induced renal failure with a cell-permeable peptide. Pediatr. Nephrol. 2011, 26, 2031–2039. [Google Scholar] [CrossRef] [PubMed]
- Tsutsuki, K.; Watanabe-Takahashi, M.; Takenaka, Y.; Kita, E.; Nishikawa, K. Identification of a Peptide-Based Neutralizer That Potently Inhibits Both Shiga Toxins 1 and 2 by Targeting Specific Receptor-Binding Regions. Infect. Immun. 2013, 81, 2133–2138. [Google Scholar] [CrossRef] [PubMed]
- Stechmann, B.; Bai, S.-K.; Gobbo, E.; Lopez, R.; Merer, G.; Pinchard, S.; Panigai, L.; Tenza, D.; Raposo, G.; Beaumelle, B.; et al. Inhibition of Retrograde Transport Protects Mice from Lethal Ricin Challenge. Cell 2010, 141, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Secher, T.; Shima, A.; Hinsinger, K.; Cintrat, J.C.; Johannes, L.; Barbier, J.; Gillet, D.; Oswald, E. Retrograde Trafficking Inhibitor of Shiga Toxins Reduces Morbidity and Mortality of Mice Infected with Enterohemorrhagic Escherichia coli. Antimicrob. Agents Chemother. 2015, 59, 5010–5013. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Linstedt, A.D. Manganese Blocks Intracellular Trafficking of Shiga Toxin and Protects Against Shiga Toxicosis. Science 2012, 335, 332–335. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Redler, B.; Linstedt, A.D. Shiga toxin–binding site for host cell receptor GPP130 reveals unexpected divergence in toxin-trafficking mechanisms. Mol. Biol. Cell 2013, 24, 2311–2318. [Google Scholar] [CrossRef] [PubMed]
- Keren, D.F.; Brown, J.E.; McDonald, R.A.; Wassef, J.S. Secretory immunoglobulin A response to Shiga toxin in rabbits: kinetics of the initial mucosal immune response and inhibition of toxicity in vitro and in vivo. Infect. Immun. 1989, 57, 1885–1889. [Google Scholar] [PubMed]
- Bitzan, M.; Poole, R.; Mehran, M.; Sicard, E.; Brockus, C.; Thuning-Roberson, C.; Rivière, M. Safety and Pharmacokinetics of Chimeric Anti-Shiga Toxin 1 and Anti-Shiga Toxin 2 Monoclonal Antibodies in Healthy Volunteers. Antimicrob. Agents Chemother. 2009, 53, 3081–3087. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.; Tzipori, S.; Sheoran, A.S. Shiga Toxin 2-Specific but Not Shiga Toxin 1-Specific Human Monoclonal Antibody Protects Piglets Challenged with Enterohemorrhagic Escherichia coli Producing Shiga Toxin 1 and Shiga Toxin 2. J. Infect. Dis. 2010, 201, 1081–1083. [Google Scholar] [CrossRef] [PubMed]
- Sheoran, A.S.; Chapman-Bonofiglio, S.; Harvey, B.R.; Mukherjee, J.; Georgiou, G.; Donohue-Rolfe, A.; Tzipori, S. Human Antibody against Shiga Toxin 2 Administered to Piglets after the Onset of Diarrhea Due to Escherichia coli O157:H7 Prevents Fatal Systemic Complications. Infect. Immun. 2005, 73, 4607–4613. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, J.M.; Mukherjee, J.; Leysath, C.E.; Debatis, M.; Ofori, K.; Baldwin, K.; Boucher, C.; Peters, R.; Beamer, G.; Sheoran, A.; et al. A Single VHH-Based Toxin-Neutralizing Agent and an Effector Antibody Protect Mice against Challenge with Shiga Toxins 1 and 2. Infect. Immun. 2013, 81, 4592–4603. [Google Scholar] [CrossRef] [PubMed]
- Luna-Gierke, R.E.; Griffin, P.M.; Gould, L.H.; Herman, K.; Bopp, C.A.; Strockbine, N.; Mody, R.K. Outbreaks of non-O157 Shiga toxin-producing Escherichia coli infection: USA. Epidemiol. Infect. 2014, 142, 2270–2280. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.-H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; LaVail, M.M.; Walter, P. IRE1 Signaling Affects Cell Fate During the Unfolded Protein Response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Scheuner, D.; Ron, D.; Pennathur, S.; Kaufman, R.J. Chop deletion reduces oxidative stress, improves β cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Investig. 2008, 118, 3378–3389. [Google Scholar] [CrossRef] [PubMed]
- Reimold, A.M.; Iwakoshi, N.N.; Manis, J.; Vallabhajosyula, P.; Szomolanyi-Tsuda, E.; Gravallese, E.M.; Friend, D.; Grusby, M.J.; Alt, F.; Glimcher, L.H. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001, 412, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wong, H.N.; Song, B.; Miller, C.N.; Scheuner, D.; Kaufman, R.J. The unfolded protein response sensor IRE1α is required at 2 distinct steps in B cell lymphopoiesis. J. Clin. Investig. 2005, 115, 268. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, A.L.; Shapiro-Shelef, M.; Iwakoshi, N.N.; Lee, A.-H.; Qian, S.-B.; Zhao, H.; Yu, X.; Yang, L.; Tan, B.K.; Rosenwald, A.; et al. XBP1, Downstream of Blimp-1, Expands the Secretory Apparatus and Other Organelles, and Increases Protein Synthesis in Plasma Cell Differentiation. Immunity 2004, 21, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Iwakoshi, N.N.; Pypaert, M.; Glimcher, L.H. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J. Exp. Med. 2007, 204, 2267–2275. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, D.; Bevan, M.J. Endoplasmic Reticulum Stress Regulator XBP-1 Contributes to Effector CD8+ T Cell Differentiation during Acute Infection. J. Immunol. Baltim. Md 1950 2008, 181, 5433. [Google Scholar] [CrossRef]
- Lee, A.-H.; Heidtman, K.; Hotamisligil, G.S.; Glimcher, L.H. Dual and opposing roles of the unfolded protein response regulated by IRE1α and XBP1 in proinsulin processing and insulin secretion. Proc. Natl. Acad. Sci. USA 2011, 108, 8885–8890. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-S.; Koo, S.; Jeong, D.; Tesh, V. Shiga Toxins as Multi-Functional Proteins: Induction of Host Cellular Stress Responses, Role in Pathogenesis and Therapeutic Applications. Toxins 2016, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Tesh, V.L. The induction of apoptosis by Shiga toxins and ricin. Curr. Top. Microbiol. Immunol. 2012, 357, 137–178. [Google Scholar] [CrossRef] [PubMed]
- Falguières, T.; Johannes, L. Shiga toxin B-subunit binds to the chaperone BiP and the nucleolar protein B23. Biol. Cell 2006, 98, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Haslam, D.B. Shiga Toxin Is Transported from the Endoplasmic Reticulum following Interaction with the Luminal Chaperone HEDJ/ERdj3. Infect. Immun. 2005, 73, 2524–2532. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-S.; Cherla, R.P.; Lentz, E.K.; Leyva-Illades, D.; Tesh, V.L. Signaling through C/EBP Homologous Protein and Death Receptor 5 and Calpain Activation Differentially Regulate THP-1 Cell Maturation-Dependent Apoptosis Induced by Shiga Toxin Type 1. Infect. Immun. 2010, 78, 3378–3391. [Google Scholar] [CrossRef] [PubMed]
- Fujii, J.; Wood, K.; Matsuda, F.; Carneiro-Filho, B.A.; Schlegel, K.H.; Yutsudo, T.; Binnington-Boyd, B.; Lingwood, C.A.; Obata, F.; Kim, K.S.; et al. Shiga Toxin 2 Causes Apoptosis in Human Brain Microvascular Endothelial Cells via C/EBP Homologous Protein. Infect. Immun. 2008, 76, 3679–3689. [Google Scholar] [CrossRef] [PubMed]
- Özcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.-H.; Iwakoshi, N.N.; Özdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic Reticulum Stress Links Obesity, Insulin Action, and Type 2 Diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Mollereau, B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 2014, 15, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Lee, A.-H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.S.; Higgins, D.E.; Schreiber, S.; Glimcher, L.H.; et al. XBP1 Links ER Stress to Intestinal Inflammation and Confers Genetic Risk for Human Inflammatory Bowel Disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.X.; Ma, J.H.; Bhatta, M.; Fliesler, S.J.; Wang, J.J. The unfolded protein response in retinal vascular diseases: Implications and therapeutic potential beyond protein folding. Prog. Retin. Eye Res. 2015, 45, 111–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Binder, P.; Fang, Q.; Wang, Z.; Xiao, W.; Liu, W.; Wang, X. Endoplasmic reticulum stress in the heart: insights into mechanisms and drug targets. Br. J. Pharmacol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Alam, G.N.; Ning, Y.; Visioli, F.; Dong, Z.; Nör, J.E.; Polverini, P.J. The Unfolded Protein Response Induces the Angiogenic Switch in Human Tumor Cells through the PERK/ATF4 Pathway. Cancer Res. 2012, 72, 5396–5406. [Google Scholar] [CrossRef] [PubMed]
- Rodvold, J.J.; Chiu, K.T.; Hiramatsu, N.; Nussbacher, J.K.; Galimberti, V.; Mahadevan, N.R.; Willert, K.; Lin, J.H.; Zanetti, M. Intercellular transmission of the unfolded protein response promotes survival and drug resistance in cancer cells. Sci. Signal 2017, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Blais, J.D.; Addison, C.L.; Edge, R.; Falls, T.; Zhao, H.; Wary, K.; Koumenis, C.; Harding, H.P.; Ron, D.; Holcik, M.; et al. Perk-Dependent Translational Regulation Promotes Tumor Cell Adaptation and Angiogenesis in Response to Hypoxic Stress. Mol. Cell. Biol. 2006, 26, 9517–9532. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.M.; Girard, M.C.; Álvarez, R.S.; Paton, A.W.; Paton, J.C.; Repetto, H.A.; Sacerdoti, F.; Ibarra, C.A. Ouabain Protects Human Renal Cells against the Cytotoxic Effects of Shiga Toxin Type 2 and Subtilase Cytotoxin. Toxins 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Engin, F.; Yermalovich, A.; Nguyen, T.; Hummasti, S.; Fu, W.; Eizirik, D.L.; Mathis, D.; Hotamisligil, G.S. Restoration of the Unfolded Protein Response in Pancreatic β Cells Protects Mice against Type 1 Diabetes. Sci. Transl. Med. 2013, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Zimmermann, E.M.; Chuang, B.; Song, B.; Nwokoye, A.; Wilkinson, J.E.; Eaton, K.A.; Kaufman, R.J. The Unfolded Protein Response and Chemical Chaperones Reduce Protein Misfolding and Colitis in Mice. Gastroenterology 2013, 144, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Kars, M.; Yang, L.; Gregor, M.F.; Mohammed, B.S.; Pietka, T.A.; Finck, B.N.; Patterson, B.W.; Horton, J.D.; Mittendorfer, B.; Hotamisligil, G.S.; et al. Tauroursodeoxycholic Acid May Improve Liver and Muscle but Not Adipose Tissue Insulin Sensitivity in Obese Men and Women. Diabetes 2010, 59, 1899–1905. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Giacca, A.; Lewis, G.F. Sodium Phenylbutyrate, a Drug With Known Capacity to Reduce Endoplasmic Reticulum Stress, Partially Alleviates Lipid-Induced Insulin Resistance and β-Cell Dysfunction in Humans. Diabetes 2011, 60, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Yusta, B.; Baggio, L.L.; Estall, J.L.; Koehler, J.A.; Holland, D.P.; Li, H.; Pipeleers, D.; Ling, Z.; Drucker, D.J. GLP-1 receptor activation improves β cell function and survival following induction of endoplasmic reticulum stress. Cell Metab. 2006, 4, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Iordanov, M.S.; Pribnow, D.; Magun, J.L.; Dinh, T.H.; Pearson, J.A.; Chen, S.L.; Magun, B.E. Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol. Cell. Biol. 1997, 17, 3373. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-R.; Lau, A.S.; Pestka, J.J. Role of Double-Stranded RNA-Activated Protein Kinase R (PKR) in Deoxynivalenol-Induced Ribotoxic Stress Response. Toxicol. Sci. 2003, 74, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.; Gray, J.S.; Li, M.; Vines, L.; Kim, J.; Pestka, J.J. Hematopoietic Cell Kinase Associates with the 40S Ribosomal Subunit and Mediates the Ribotoxic Stress Response to Deoxynivalenol in Mononuclear Phagocytes. Toxicol. Sci. 2010, 115, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Foster, G.H.; Tesh, V.L. Shiga toxin 1-induced activation of c-Jun NH2-terminal kinase and p38 in the human monocytic cell line THP-1: possible involvement in the production of TNF-α. J. Leukoc. Biol. 2002, 71, 107–114. [Google Scholar] [PubMed]
- Brandelli, J.R.; Griener, T.P.; Laing, A.; Mulvey, G.; Armstrong, G.D. The Effects of Shiga Toxin 1, 2 and Their Subunits on Cytokine and Chemokine Expression by Human Macrophage-Like THP-1 Cells. Toxins 2015, 7, 4054–4066. [Google Scholar] [CrossRef] [PubMed]
- Moazzezy, N.; Oloomi, M.; Bouzari, S. Effect of Shiga Toxin and Its Subunits on Cytokine Induction in Different Cell Lines. Int. J. Mol. Cell. Med. 2014, 3, 108. [Google Scholar] [PubMed]
- Smith, W.E.; Kane, A.V.; Campbell, S.T.; Acheson, D.W.K.; Cochran, B.H.; Thorpe, C.M. Shiga Toxin 1 Triggers a Ribotoxic Stress Response Leading to p38 and JNK Activation and Induction of Apoptosis in Intestinal Epithelial Cells. Infect. Immun. 2003, 71, 1497–1504. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, D.M.; Rogers, T.J.; Kane, A.; Paton, A.W.; Paton, J.C.; Thorpe, C.M. Shiga Toxin 2 and Flagellin from Shiga-Toxigenic Escherichia coli Superinduce Interleukin-8 through Synergistic Effects on Host Stress-Activated Protein Kinase Activation. Infect. Immun. 2010, 78, 2984–2994. [Google Scholar] [CrossRef] [PubMed]
- Bunyard, P.; Handley, M.; Pollara, G.; Rutault, K.; Wood, I.; Chaudry, M.; Alderman, C.; Foreman, J.; Katz, D.R.; Chain, B.M. Ribotoxic stress activates p38 and JNK kinases and modulates the antigen-presenting activity of dendritic cells. Mol. Immunol. 2003, 39, 815–827. [Google Scholar] [CrossRef]
- Eisenhauer, P.B.; Jacewicz, M.S.; Conn, K.J.; Koul, O.; Wells, J.M.; Fine, R.E.; Newburg, D.S. Escherichia coli Shiga toxin 1 and TNF-α induce cytokine release by human cerebral microvascular endothelial cells. Microb. Pathog. 2004, 36, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Ramegowda, B.; Samuel, J.E.; Tesh, V.L. Interaction of Shiga Toxins with Human Brain Microvascular Endothelial Cells: Cytokines as Sensitizing Agents. J. Infect. Dis. 1999, 180, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Petruzziello-Pellegrini, T.N.; Yuen, D.A.; Page, A.V.; Patel, S.; Soltyk, A.M.; Matouk, C.C.; Wong, D.K.; Turgeon, P.J.; Fish, J.E.; Ho, J.J.D.; et al. The CXCR4/CXCR7/SDF-1 pathway contributes to the pathogenesis of Shiga toxin–associated hemolytic uremic syndrome in humans and mice. J. Clin. Investig. 2012, 122, 759. [Google Scholar] [CrossRef] [PubMed]
- Stearns-Kurosawa, D.J.; Collins, V.; Freeman, S.; Tesh, V.L.; Kurosawa, S. Distinct physiologic and inflammatory responses elicited in baboons after challenge with Shiga toxin type 1 or 2 from enterohemorrhagic Escherichia coli. Infect. Immun. 2010, 78, 2497–2504. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, D.M.; Wong, J.; Mantis, N.J.; Magun, B.E.; Leong, J.M.; Thorpe, C.M. A Novel Zak Knockout Mouse with a Defective Ribotoxic Stress Response. Toxins 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.M.; Thorpe, C.M.; Ahluwalia, A.; Rogers, A.B.; Obata, F.; Vozenilek, A.; Kolling, G.L.; Kane, A.V.; Magun, B.E.; Jandhyala, D.M. Shiga toxin 2-induced intestinal pathology in infant rabbits is A-subunit dependent and responsive to the tyrosine kinase and potential ZAK inhibitor imatinib. Front. Cell. Infect. Microbiol. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Karpman, D.; Andreasson, A.; Thysell, H.; Kaplan, B.S.; Svanborg, C. Cytokines in childhood hemolytic uremic syndrome and thrombotic thrombocytopenic purpura. Pediatr. Nephrol. Berl. Ger. 1995, 9, 694–699. [Google Scholar] [CrossRef]
- Hagel, C.; Krasemann, S.; Löffler, J.; Püschel, K.; Magnus, T.; Glatzel, M. Upregulation of Shiga Toxin Receptor CD77/Gb3 and Interleukin-1β Expression in the Brain of EHEC Patients with Hemolytic Uremic Syndrome and Neurologic Symptoms. Brain Pathol. 2015, 25, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Lopez, E.L.; Contrini, M.M.; Devoto, S.; De Rosa, M.F.P.; Grana, M.G.; Genero, M.H.; Canepa, C.; Gomez, H.F.; Cleary, T.G. Tumor necrosis factor concentrations in hemolytic uremic syndrome patients and children with bloody diarrhea in Argentina. Pediatr. Infect. Dis. J. 1995, 14, 594–598. [Google Scholar] [CrossRef] [PubMed]
- Alves-Rosa, F.; Beigier-Bompadre, M.; Fernández, G.; Barrionuevo, P.; Mari, L.; Palermo, M.; Isturiz, M. Tolerance to lipopolysaccharide (LPS) regulates the endotoxin effects on Shiga toxin-2 lethality. Immunol. Lett. 2001, 76, 125–131. [Google Scholar] [CrossRef]
- Pinto, A.; Cangelosi, A.; Geoghegan, P.A.; Goldstein, J. Dexamethasone prevents motor deficits and neurovascular damage produced by shiga toxin 2 and lipopolysaccharide in the mouse striatum. Neuroscience 2017, 344, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-M.; Ou, Z.L.; Gondaira, F.; Ohmura, M.; Kojio, S.; Yamamoto, T. Protective effect of anisodamine against Shiga toxin-1: Inhibition of cytokine production and increase in the survival of mice. J. Lab. Clin. Med. 2001, 137, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Keepers, T.R.; Gross, L.K.; Obrig, T.G. Monocyte Chemoattractant Protein 1, Macrophage Inflammatory Protein 1α, and RANTES Recruit Macrophages to the Kidney in a Mouse Model of Hemolytic-Uremic Syndrome. Infect. Immun. 2007, 75, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Isogai, E.; Isogai, H.; Hirose, K.; Kubota, T.; Kimura, K.; Fujii, N.; Hayashi, S.; Takeshi, K.; Oguma, K. Therapeutic effect of anti-TNF-α antibody and levofloxacin (LVFX) in a mouse model of enterohemorrhagic Escherichia coli O157 infection. Comp. Immunol. Microbiol. Infect. Dis. 2001, 24, 217–231. [Google Scholar] [CrossRef]
- Argollo, M.; Fiorino, G.; Hindryck, P.; Peyrin-Biroulet, L.; Danese, S. Novel therapeutic targets for inflammatory bowel disease. J. Autoimmun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Mateen, S.; Zafar, A.; Moin, S.; Khan, A.Q.; Zubair, S. Understanding the role of cytokines in the pathogenesis of rheumatoid arthritis. Clin. Chim. Acta 2016, 455, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Marcuzzi, A.; Piscianz, E.; Valencic, E.; Monasta, L.; Vecchi Brumatti, L.; Tommasini, A. To Extinguish the Fire from Outside the Cell or to Shutdown the Gas Valve Inside? Novel Trends in Anti-Inflammatory Therapies. Int. J. Mol. Sci. 2015, 16, 21277–21293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandrup-Poulsen, T. Interleukin-1 Antagonists and Other Cytokine Blockade Strategies for Type 1 Diabetes. Rev. Diabet. Stud. RDS 2012, 9, 338–347. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Therapeutic | Drug Class | Target | Mechanism of Action | Animal Models Tested | Clinical Trials Completed | References |
|---|---|---|---|---|---|---|
| Anti-sera | Polyclonal antibodies | STX, STX2 | Circulating toxin neutralization | Pig, rabbit | None | [49,67] |
| Urtoxezumab® | Humanized murine monoclonal antibody | STX2 | Circulating toxin neutralization | Rodent, NHP | Phase II | [50,53] |
| cαSTX1 and cαSTX2 | Humanized murine monoclonal antibody | STX1, STX2 | Circulating toxin neutralization | Rodent | Phase I | [54,68] |
| Murine anti-STX2 | Murine monoclonal antibody | STX2 | Circulating toxin neutralization | Rodent | None | [52] |
| Anti-STX antibodies (various clones) | Human monoclonal antibody | STX1, STX2 | Circulating toxin neutralization | Rodent, pig | None | [51,69,70] |
| Camelid anti-STX oligomers | VHH-based neutralizing agent | STX1, STX2 | Circulating toxin neutralization | Rodent | None | [55,71] |
| Adenoviral anti-STX2 construct | VHH-based neutralizing agent | STX2 | Circulating toxin neutralization | Rodent, pig | None | [56] |
| Tetravalent peptides | Gb3 analogs | STX1, STX2 | Circulating toxin neutralization | Rodent, non-human primate | None | [59,60,61,62] |
| Synsorb-Pk® | Silicon dioxide-Gb3 construct | STX1, STX2 | Gastrointestinal toxin neutralization | None | Phase II (failed) | [57,58] |
| Retro 1 and Retro 2 | Small molecule inhibitors | STX1, STX2 | Retrograde trafficking inhibitor | Rodent | None | [63,64] |
| Manganese | Enzyme cofactor | STX1 | Retrograde trafficking inhibitor | Rodent | None | [65] |
| Disease | Cells Type Affected | Outcome | Model System(s) | Reference |
|---|---|---|---|---|
| Shiga toxicosis following STEC infection | Leukocytes, endothelial cells, renal epithelium, gastrointestinal epithelium | Hemolytic uremic syndrome? Inflammatory cytokine secretion? | Rodent, human monocyte, renal epithelial, and endothelial cells in vitro | [34,44,46,89,90] |
| Diabetes mellitus | Pancreatic beta cells | Loss of insulin production | Rodent | [91,92] |
| Obesity | Hepatocytes | Hepatic lipidosis, insulin resistance | Rodent, various hepatocyte cell lines in vitro | [91,92] |
| Inflammatory Bowel Disease | Intestinal Paneth and goblet cells | Loss of Paneth cells, gastrointestinal inflammation | Rodent | [94,95] |
| Neurodegenerative Diseases | Neurons | Neuron dysfunction and degeneration | Rodent | [93] |
| Vascular retinopathies | Retinal endothelial and pigmented epithelial cells | Aberrant angiogenesis | Rodent, human retinal endothelial cells and pigmented retinal epithelial cells in vitro | [96] |
| Cardiac disease | Cardiomyocytes | Cardiac hypertrophy, arrhythmias, cardiac fibrosis | Rodent, rabbit, human cardiomyocytes in vitro | [97] |
| Neoplasia | Malignant cells | Inflammatory cytokine secretion, angiogenesis, tumor survival | Human-mouse xenografts, neoplastic cells in vitro | [98,99,100] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hall, G.; Kurosawa, S.; Stearns-Kurosawa, D.J. Shiga Toxin Therapeutics: Beyond Neutralization. Toxins 2017, 9, 291. https://doi.org/10.3390/toxins9090291
Hall G, Kurosawa S, Stearns-Kurosawa DJ. Shiga Toxin Therapeutics: Beyond Neutralization. Toxins. 2017; 9(9):291. https://doi.org/10.3390/toxins9090291
Chicago/Turabian StyleHall, Gregory, Shinichiro Kurosawa, and Deborah J. Stearns-Kurosawa. 2017. "Shiga Toxin Therapeutics: Beyond Neutralization" Toxins 9, no. 9: 291. https://doi.org/10.3390/toxins9090291
APA StyleHall, G., Kurosawa, S., & Stearns-Kurosawa, D. J. (2017). Shiga Toxin Therapeutics: Beyond Neutralization. Toxins, 9(9), 291. https://doi.org/10.3390/toxins9090291