Biometric Image Analysis for Quantitation of Dividing Platelets


Abstract
:1. Introduction
1.1. Background
1.2. The Problem of Low Platelet Count
1.3. Platelet Division
2. Materials and Methods
2.1. Platelet Isolation from Blood
2.2. Platelet Count before and after Suspension Culture Measured Using an Automated Coulter Counter
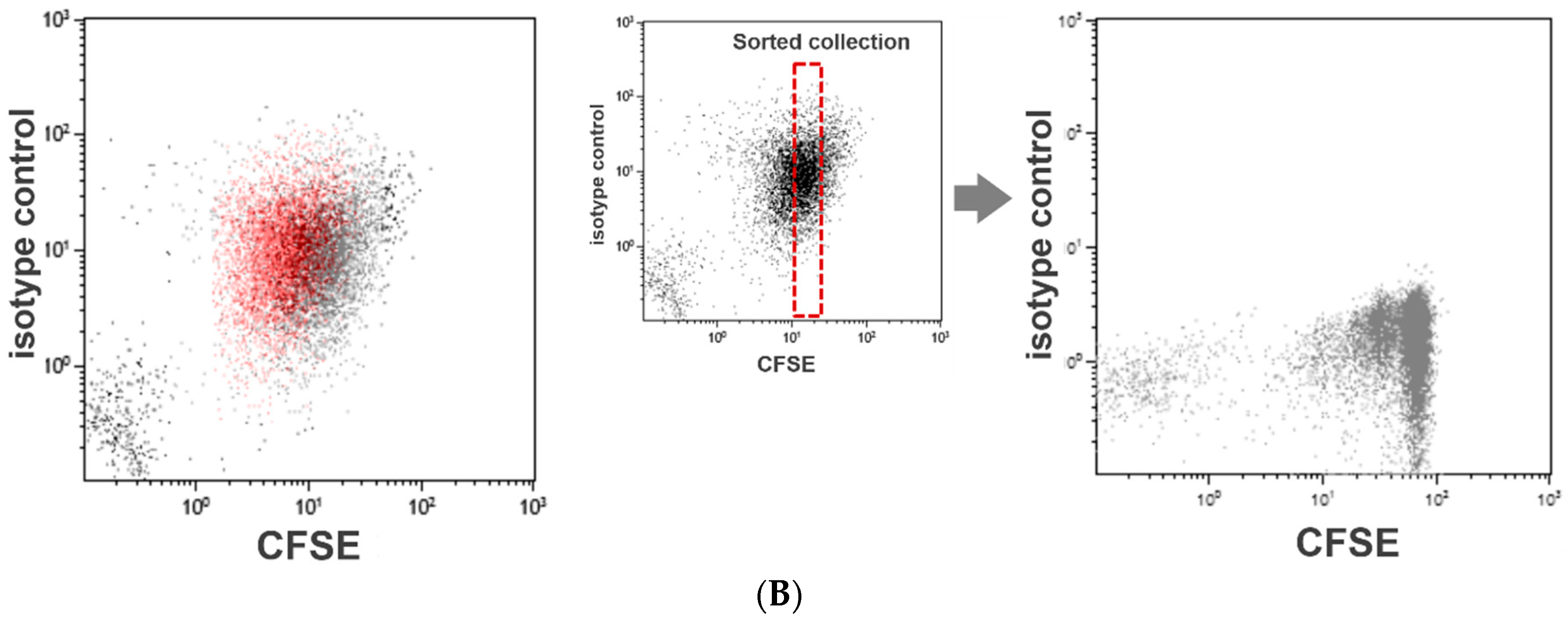
2.3. CFSE Dilution Assay to Assess Platelet Division
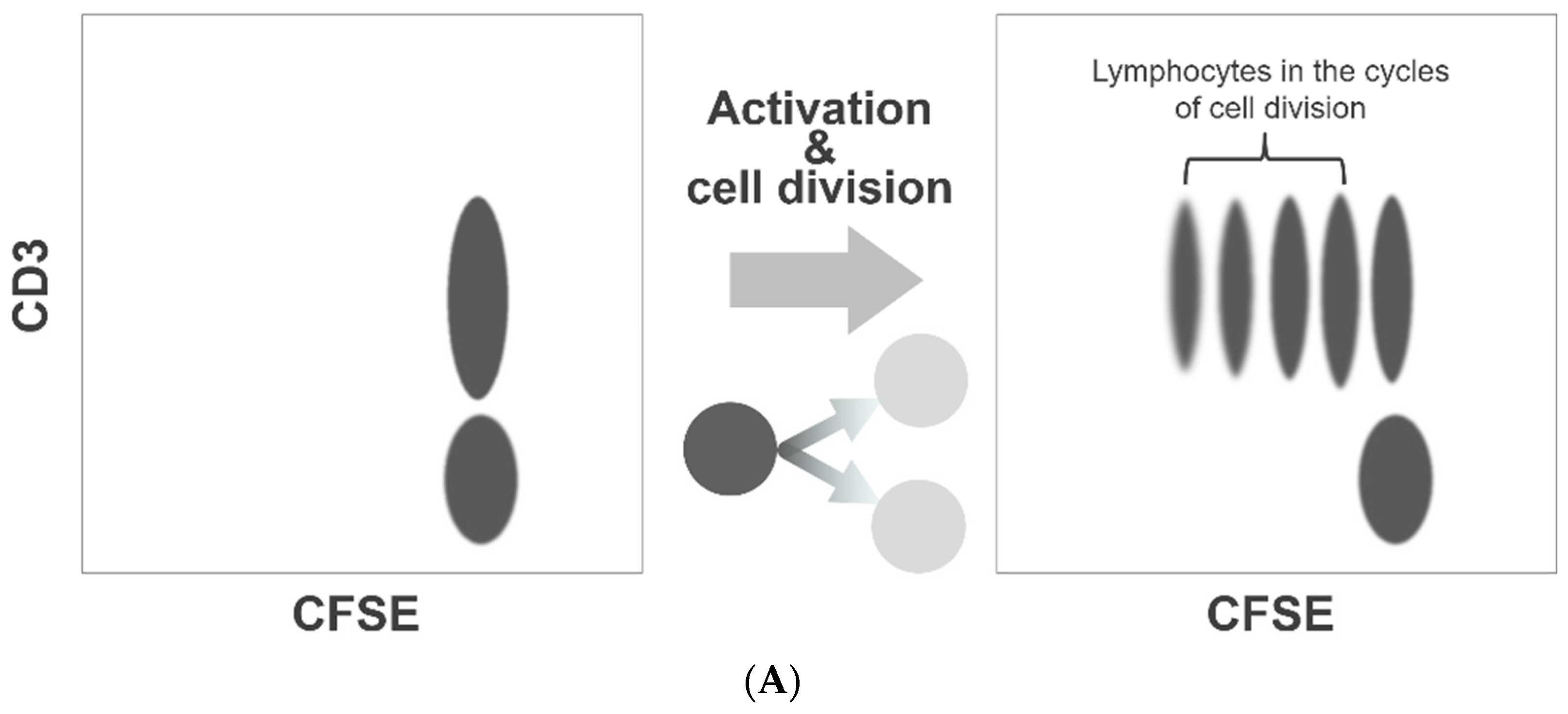
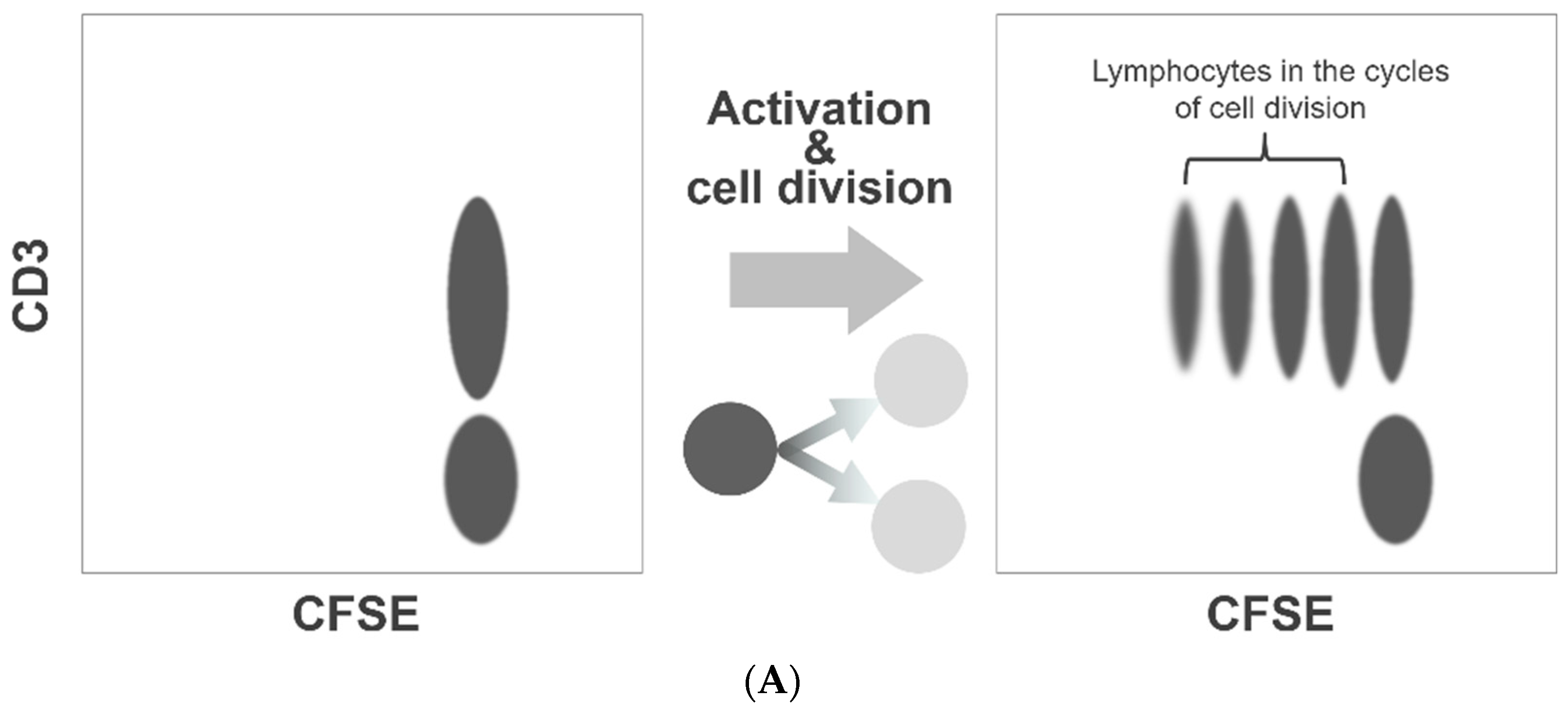
2.3.1. Principle
2.3.2. Procedure for Assessment of Platelet Division
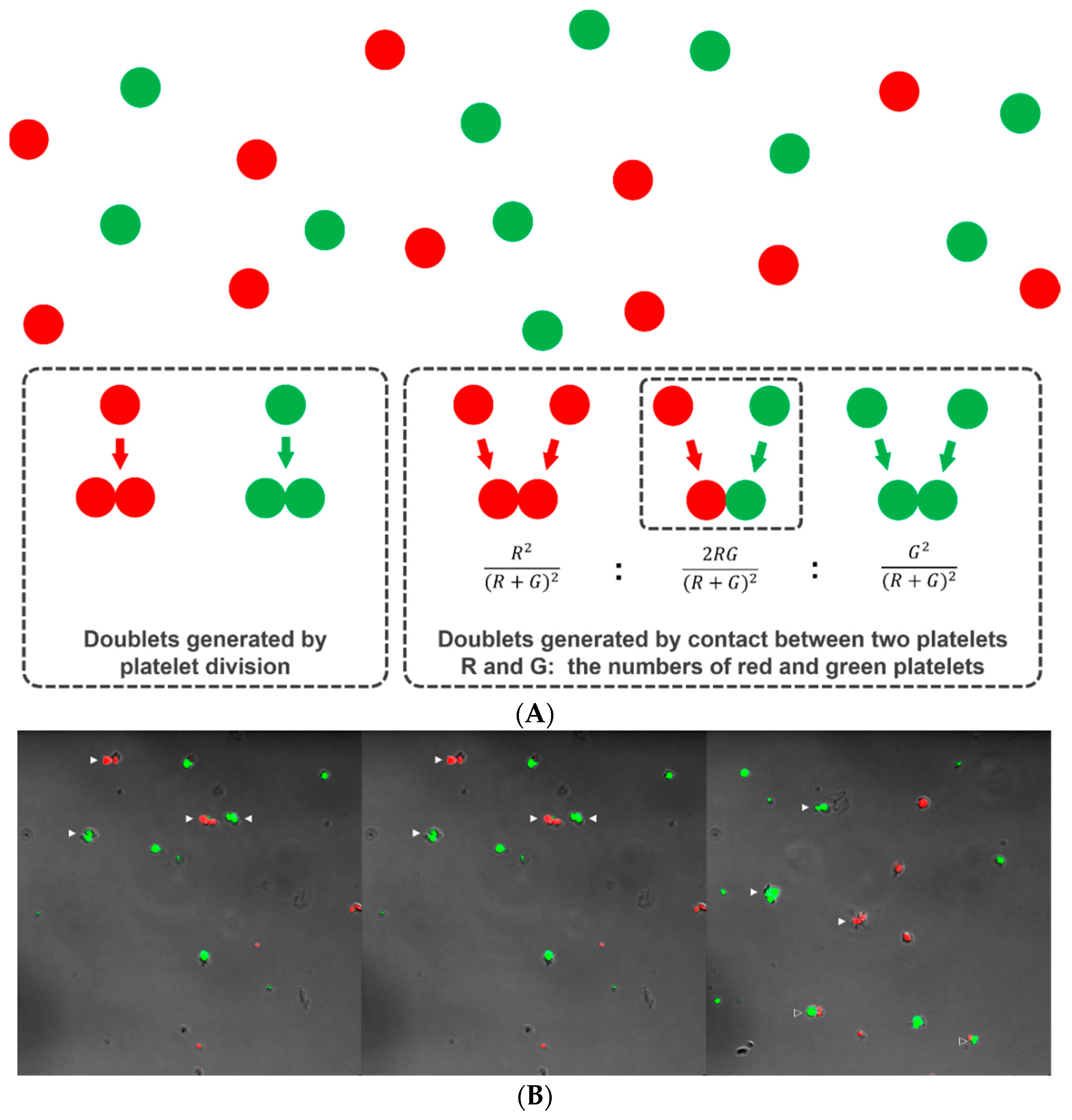
2.4. Differential Counting of Platelet Doublets
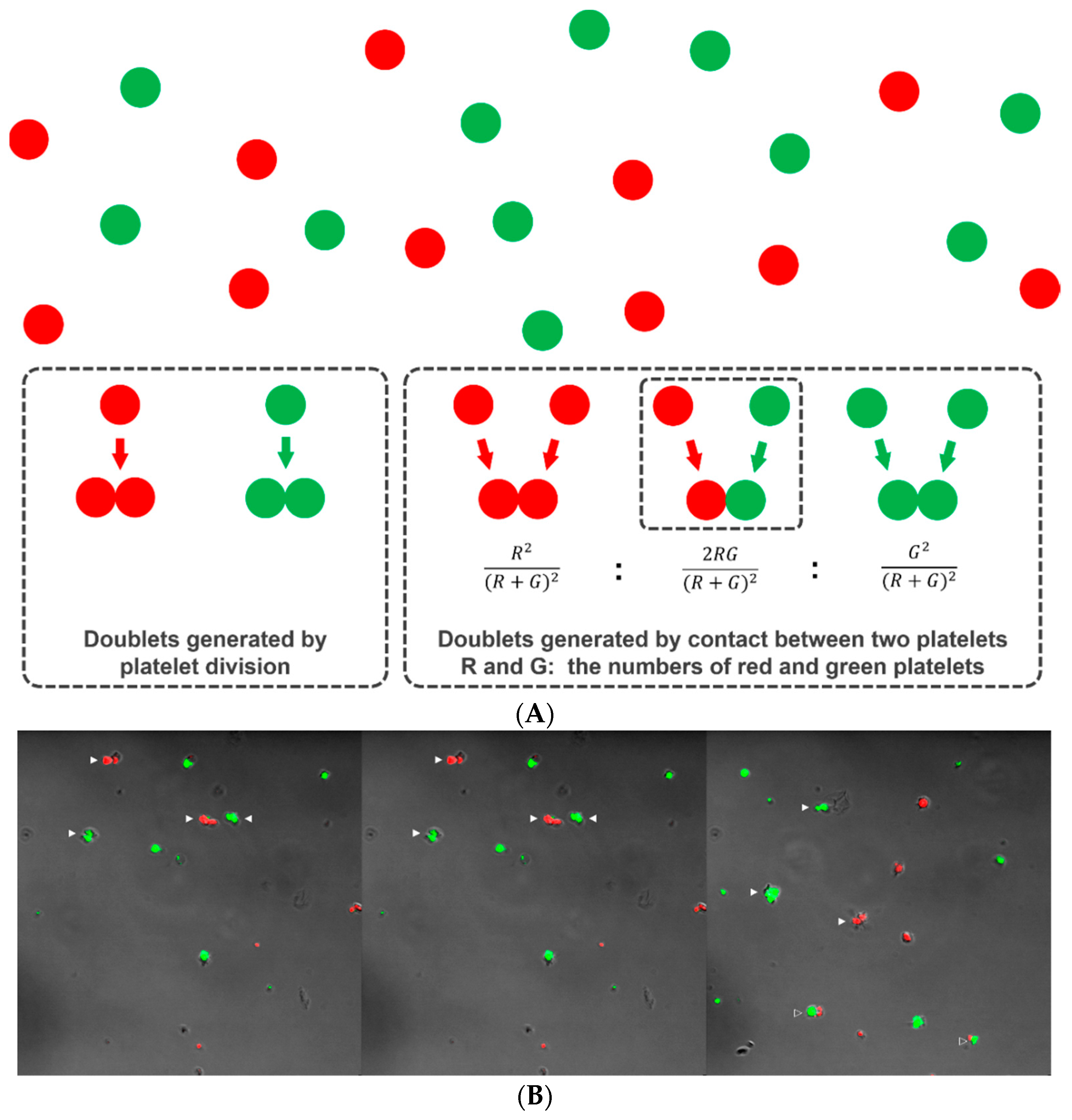
2.4.1. Principle
2.4.2. Demonstration of Platelet Division by Differential Doublet Counting
3. Results and Discussion
3.1. Platelet Counting Using an Automated Hematology Analyzer Based on Coulter Counting
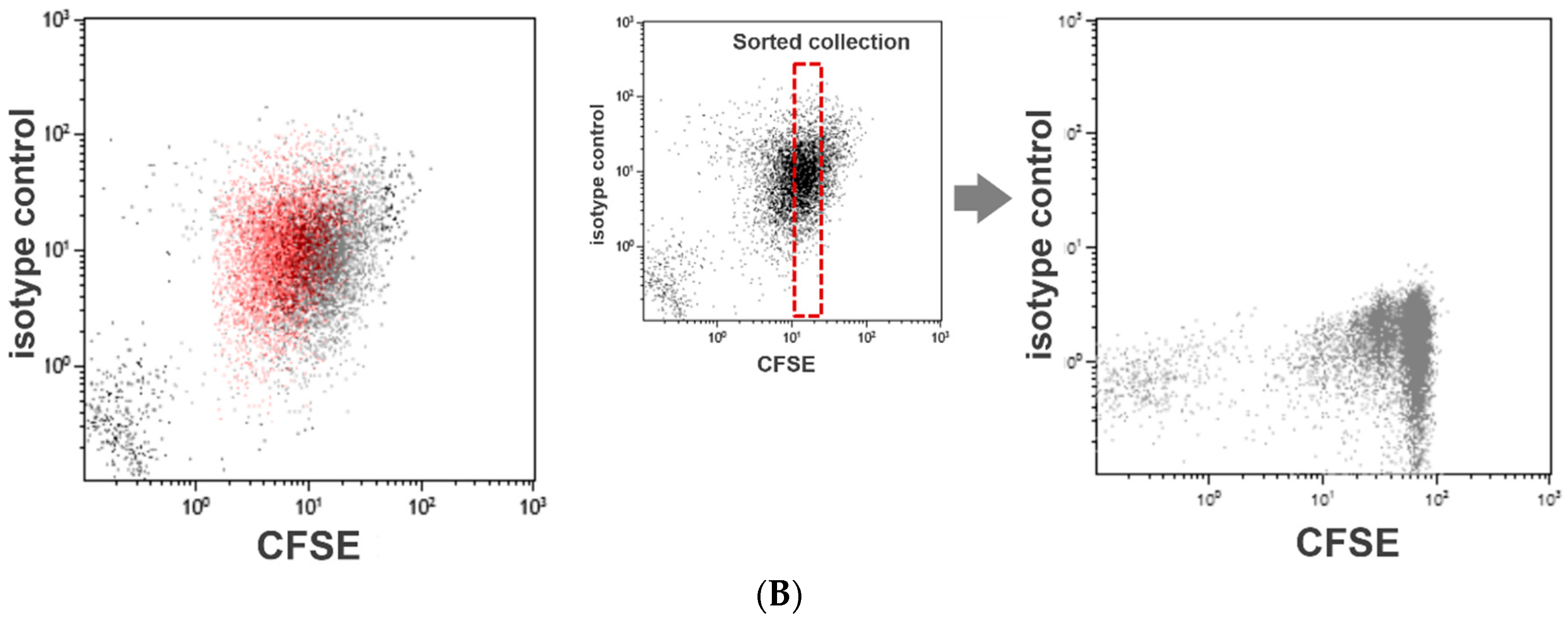
3.2. Assessment of Platelet Division by CFSE Dilution Assay
3.3. Assessment of Platelet Division by Differential Doublet Counting
3.3.1. Demonstration of Platelet Division by Differential Doublet Counting
3.3.2. Cytoskeletal Rearrangement in Platelet Division
3.4. Derivation of the Dividing Fraction of Platelets from Differential Doublet Counting
3.4.1. Derivation of the Formulae for Platelet Division Fraction
3.4.2. Calculated Platelet Division Fraction
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jurk, K.; Kehrel, B.E. Platelets: Physiology and biochemistry. Semin. Thromb. Hemost. 2005, 31, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Smyth, S.S.; McEver, R.P.; Weyrich, A.S.; Morrell, C.N.; Hoffman, M.R.; Arepally, G.M.; French, P.A.; Dauerman, H.L.; Becker, R.C. Platelet functions beyond hemostasis. J. Thromb. Haemost. 2009, 7, 1759–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, R.K.; Berndt, M.C. Platelet physiology and thrombosis. Thromb. Res. 2004, 114, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Nuyttens, B.P.; Thijs, T.; Deckmyn, H.; Broos, K. Platelet adhesion to collagen. Thromb. Res. 2011, 127, S26–S29. [Google Scholar] [CrossRef]
- Levy, J.H.; Rossaint, R.; Zacharowski, K.; Spahn, D.R. What is the evidence for platelet transfusion in perioperative settings? Vox Sang. 2017, 112, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Estcourt, L.J.; Malouf, R.; Doree, C.; Trivella, M.; Hopewell, S.; Birchall, J. Prophylactic platelet transfusions prior to surgery for people with a low platelet count. Cochrane Database Syst. Rev. 2018, 9. [Google Scholar] [CrossRef]
- Kaufman, R.M.; Djulbegovic, B.; Gernsheimer, T.; Kleinman, S.; Tinmouth, A.T.; Capocelli, K.E.; Cipolle, M.D.; Cohn, C.S.; Fung, M.K.; Grossman, B.J.; et al. Platelet transfusion: A clinical practice guideline from the AABB. Ann. Intern. Med. 2015, 162, 205–213. [Google Scholar] [CrossRef]
- Rodeghiero, F. Idiopathic thrombocytopenic purpura: An old disease revisited in the era of evidence-based medicine. Haematologica 2003, 88, 1081–1087. [Google Scholar]
- Gernsheimer, T. Chronic idiopathic thrombocytopenic purpura: Mechanisms of pathogenesis. Oncologist 2009, 14, 12–21. [Google Scholar] [CrossRef]
- Cooper, N. State of the art—How I manage immune thrombocytopenia. Br. J. Haematol. 2017, 177, 39–54. [Google Scholar] [CrossRef]
- Stasi, R. Eltrombopag for the treatment of idiopathic thrombocytopenic purpura. Expert Rev. Hematol. 2008, 1, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Kuhne, T.; Imbach, P. Eltrombopag: An update on the novel, non-peptide thrombopoietin receptor agonist for the treatment of immune thrombocytopenia. Ann. Hematol. 2010, 89, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Cuker, A. Toxicities of the thrombopoietic growth factors. Semin. Hematol. 2010, 47, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Machlus, K.R.; Thon, J.N.; Italiano, J.E., Jr. Interpreting the developmental dance of the megakaryocyte: A review of the cellular and molecular processes mediating platelet formation. Br. J. Haematol. 2014, 165, 227–236. [Google Scholar] [CrossRef]
- Schwertz, H.; Koster, S.; Kahr, W.H.; Michetti, N.; Kraemer, B.F.; Weitz, D.A.; Blaylock, R.C.; Kraiss, L.W.; Greinacher, A.; Zimmerman, G.A.; et al. Anucleate platelets generate progeny. Blood 2010, 115, 3801–3809. [Google Scholar] [CrossRef] [Green Version]
- Thon, J.N.; Montalvo, A.; Patel-Hett, S.; Devine, M.T.; Richardson, J.L.; Ehrlicher, A.; Larson, M.K.; Hoffmeister, K.; Hartwig, J.H.; Italiano, J.E., Jr. Cytoskeletal mechanics of proplatelet maturation and platelet release. J. Cell Biol. 2010, 191, 861–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Q.; Palliser, D.; Eisen, H.N.; Chen, J. Homeostatic T cell proliferation in a T cell-dendritic cell coculture system. Proc. Natl. Acad. Sci. USA 2002, 99, 2983–2988. [Google Scholar] [CrossRef]
- Hussain, K.; Hargreaves, C.E.; Roghanian, A.; Oldham, R.J.; Chan, H.T.; Mockridge, C.I.; Chowdhury, F.; Frendeus, B.; Harper, K.S.; Strefford, J.C.; et al. Upregulation of FcγRIIb on monocytes is necessary to promote the superagonist activity of TGN1412. Blood 2015, 125, 102–110. [Google Scholar] [CrossRef]
- De Cuyper, I.M.; Meinders, M.; van de Vijver, E.; de Korte, D.; Porcelijn, L.; de Haas, M.; Eble, J.A.; Seeger, K.; Rutella, S.; Pagliara, D.; et al. A novel flow cytometry-based platelet aggregation assay. Blood 2013, 121, e70–e80. [Google Scholar] [CrossRef] [Green Version]
- Dunois-Larde, C.; Capron, C.; Fichelson, S.; Bauer, T.; Cramer-Borde, E.; Baruch, D. Exposure of human megakaryocytes to high shear rates accelerates platelet production. Blood 2009, 114, 1875–1883. [Google Scholar] [CrossRef] [Green Version]
- Bender, M.; Thon, J.N.; Ehrlicher, A.J.; Wu, S.; Mazutis, L.; Deschmann, E.; Sola-Visner, M.; Italiano, J.E.; Hartwig, J.H. Microtubule sliding drives proplatelet elongation and is dependent on cytoplasmic dynein. Blood 2015, 125, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.R.; Richardson, J.L.; Schulze, H.; Kahle, E.; Galjart, N.; Drabek, K.; Shivdasani, R.A.; Hartwig, J.H.; Italiano, J.E., Jr. Differential roles of microtubule assembly and sliding in proplatelet formation by megakaryocytes. Blood 2005, 106, 4076–4085. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment Number | Initial | 6 Hours | 20 Hours | |||
|---|---|---|---|---|---|---|
| High MFI | Low MFI | High MFI | Low MFI | High MFI | Low MFI | |
| Sort-1 | 15.5 | 9.4 | 41.4 | 43.4 | 90.6 | 97.0 |
| Sort-2 | 14.3 | 7.9 | 39.7 | 41.8 | 87.3 | 86.5 |
| Sort-3 | 10.9 | 6.3 | 27.5 | 27.7 | 81.1 | 34.5 |
| Sort-4 | 16.4 | 10.9 | 64.5 | 37.7 | 76.4 | 62.4 |
| Sort-5 | 12.0 | 11.1 | 70.4 | 24.6 | 83.3 | 48.1 |
| Sort-6 | 38.5 | 20.3 | 93.8 | 96.5 | ||
| Sort-7 | 15.6 | 11.4 | 89.5 | 70.7 | ||
| Sort-8 | 22.1 | 45.3 | 80.4 | 90.0 | ||
| Sort-9 | 15.6 | 11.8 | 60.6 | 68.8 | ||
| Sort-10 | 14.3 | 8.0 | 91.7 | 67.5 | ||
| Sort-11 | 12.7 | 3.9 | 95.9 | 92.3 | ||
| Sort-12 | 8.2 | 3.5 | 96.0 | 91.3 | ||
| 16.3 ± 7.8 | 12.5 ± 11.2 | 48.7 ± 18.1 | 35.0 ± 8.4 | 85.6 ± 10.1 | 75.5 ± 20.2 | |
| P = 0.81 * | P = 0.15 * | |||||
| Experiment Number (MethoCult Medium Culture) | |||||||
|---|---|---|---|---|---|---|---|
| MCX1 | MCX2 | MCX3 | MCX4 | MCX5 | MCX6 | ||
| Initial Doublet %* (Expected†) | R● | 67 | 83 | 407 | 125 | 114 | 129 |
| G● | 60 | 77 | 448 | 104 | 110 | 125 | |
| RR●● | 3 27.3 * (27.8 †) | 3 21.4 * (26.9 †) | 11 20.0 * (22.7 †) | 7 21.2 * (29.8 †) | 4 11.1 * (25.9 †) | 5 14.7 * (25.8 †) | |
| GG●● | 3 27.3 * (22.3 †) | 4 28.6 * (23.2 †) | 12 21.8 * (27.5 †) | 6 18.2 * (20.6 †) | 5 13.9 * (24.1 †) | 4 11.8 * (24.2 †) | |
| RG●● | 5 45.5 * (49.8 †) | 7 50.0 * (49.9 †) | 32 58.2 * (49.9 †) | 20 60.6 * (49.6 †) | 27 75.0 * (50.0 †) | 25 73.5 * (50.0 †) | |
| P‡ | 0.39 | 0.50 | 0.89 | 0.90 | 1.00 | 1.00 | |
| 6-HourCulture Doublet %* (Expected†) | R● | 171 | 124 | 157 | 132 | 126 | 122 |
| G● | 167 | 134 | 151 | 127 | 127 | 103 | |
| RR●● | 28 48.3 (25.6 †) | 22 39.1 (26.0 †) | 18 39.1 (26.0 †) | 24 49.0 (26.0 †) | 19 44.2 (24.8 †) | 19 47.5 (29.4 †) | |
| GG●● | 22 37.9 (24.4 †) | 24 41.3 (24.0 †) | 19 41.3 (24.0 †) | 16 32.7 (24.0 †) | 15 34.9 (25.2 †) | 14 35.0 (21.0 †) | |
| RG●● | 8 13.8 (50.0 †) | 8 19.6 (50.0 †) | 9 19.6 (50.0 †) | 9 18.4 (50.0 †) | 9 20.9 (50.0 †) | 7 17.5 (49.6 †) | |
| P‡ | <0.01 (1.76 × 10−8) | <0.01 (1.23 × 10−7) | <0.01 (1.85 × 10−5) | <0.01 (4.80 × 10−6) | <0.01 (6.88 × 10−5) | <0.01 (2.39 × 10−5) | |
| Mock Treatment | Cytoskeletal Treatment | |||||||
|---|---|---|---|---|---|---|---|---|
| Count | Doublet %(Expected) | Total | Doublet %(Expected) | |||||
| Taxol | R● | 157 | P < 0.01 ‡ (4.62 × 10−5) | 164 | P = 0.57 ‡ | |||
| G● | 149 | 144 | ||||||
| RR●● | 8 | 44.7 * (28.4 †) | 21 | 25.8 * (26.3 †) | ||||
| GG●● | 7 | 34.0 * (21.9 †) | 16 | 22.6 * (23.7 †) | ||||
| RG●● | 16 | 21.3 * (49.8 †) | 10 | 51.6 * (50.0 †) | ||||
| Nocodazole | R● | 149 | P < 0.01 ‡ (1.97 × 10−5) | 168 | P = 0.67 ‡ | |||
| G● | 146 | 166 | ||||||
| RR●● | 11 | 37.5 * (25.3 †) | 15 | 24.4 * (25.5 †) | ||||
| GG●● | 10 | 45.0 * (24.7 †) | 18 | 22.2 * (24.5 †) | ||||
| RG●● | 24 | 17.5 * (50.0 †) | 7 | 53.3 * (50.0 †) | ||||
| Cytochalasin-D | R● | 346 | P < 0.01 ‡ (3.03 × 10−6) | 301 | P = 0.61 ‡ | |||
| G● | 313 | 281 | ||||||
| RR●● | 15 | 39.6 * (26.7 †) | 21 | 26.8 * (27.6 †) | ||||
| GG●● | 12 | 41.5 * (23.3 †) | 22 | 21.4 * (22.6 †) | ||||
| RG●● | 29 | 18.9 * (49.9 †) | 10 | 51.8 * (49.9 †) | ||||
| Fraction of Division | Experiment Number (MethoCult Medium Culture) | ||||||
|---|---|---|---|---|---|---|---|
| MCX1 | MCX2 | MCX3 | MCX4 | MCX5 | MCX6 | ||
| Initial | Fr (95 % CI*) | 0.8% (−5.3–6.8) | −1.0% (−6.3–4.2) | −0.8% (−2.8–1.1) | −4.0% (−9.6–1.5) | −8.2% (−13.1–-3.3) | −5.7% (−10.4–-1.1) |
| Fg (95 % CI*) | 0.8% (−5.3–6.8) | 1.1% (−4.6–7.1) | −1.3% (−3.3–0.7) | −2.0% (−7.6–3.5) | −6.6% (−11.8–-1.4) | −6.1% (−10.5–-1.8) | |
| 6-HourCulture | Fr (95 % CI*) | 16.4% (9.5–23.3) | 17.3% (8.8–25.8) | 9.2% (3.2–15.3) | 17.5% (9.1–25.9) | 13.2% (5.4–21.0) | 14.0% (5.8–22.2) |
| Fg (95 % CI*) | 12.0% (5.8–18.3) | 17.2% (8.8–25.6) | 10.8% (4.4–17.2) | 9.7% (2.6–16.8) | 8.8% (1.9–15.7) | 11.8% (3.8–19.8) | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.-J.; Song, Y.; Song, J. Biometric Image Analysis for Quantitation of Dividing Platelets. Micromachines 2019, 10, 1. https://doi.org/10.3390/mi10010001
Kim H-J, Song Y, Song J. Biometric Image Analysis for Quantitation of Dividing Platelets. Micromachines. 2019; 10(1):1. https://doi.org/10.3390/mi10010001
Chicago/Turabian StyleKim, Hyun-Jeong, Yejin Song, and Jaewoo Song. 2019. "Biometric Image Analysis for Quantitation of Dividing Platelets" Micromachines 10, no. 1: 1. https://doi.org/10.3390/mi10010001
APA StyleKim, H.-J., Song, Y., & Song, J. (2019). Biometric Image Analysis for Quantitation of Dividing Platelets. Micromachines, 10(1), 1. https://doi.org/10.3390/mi10010001