Multiplexed PCR-Free Detection of MicroRNAs in Single Cancer Cells Using a DNA-Barcoded Microtrough Array Chip



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
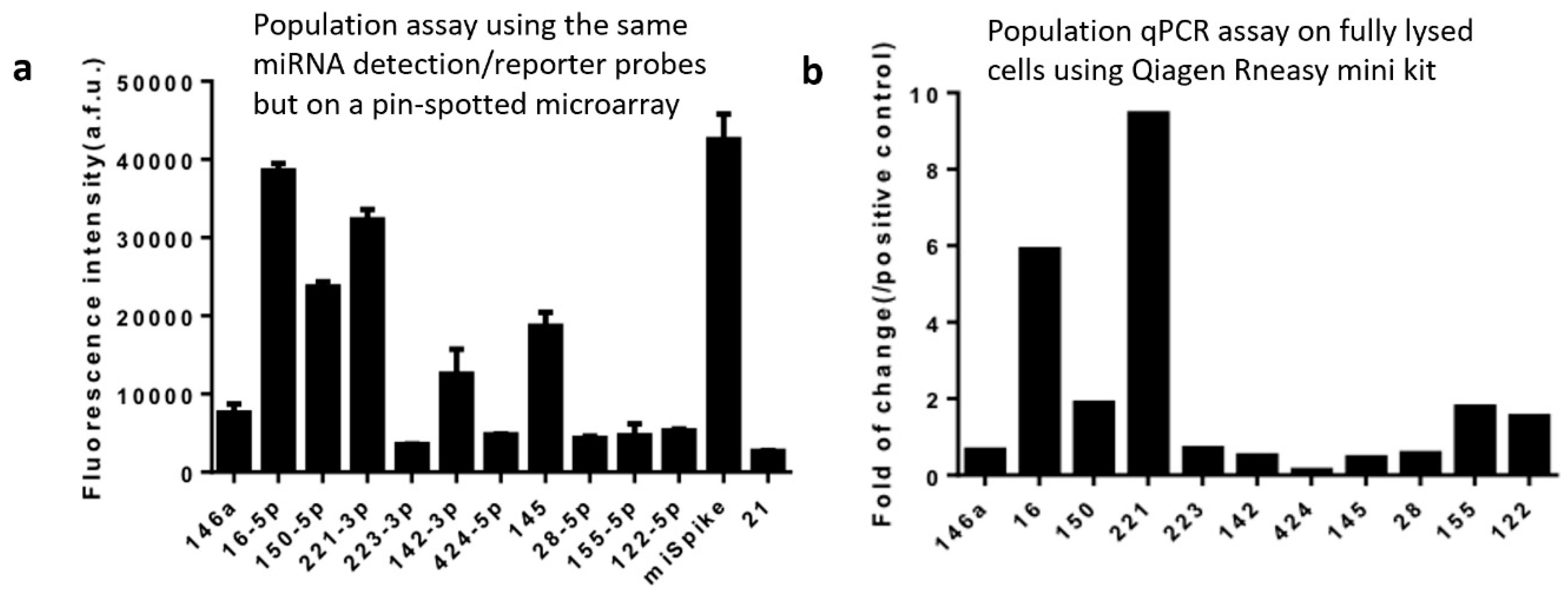{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Fabrication of Flow Patterning Device for Creating DNA Barcode Microarrays
2.2. Fabrication of Sub-Nanoliter Microtrough Array Chips
2.3. Flow Patterning of DNA Oligomers for Detecting The Reporter Probes
2.4. Design of MicroRNA Capture and Reporter Probes
2.5. Population Level Multiplexed MicroRNA Assay
2.6. Single-Cell Multiplexed MicroRNA Assay
2.7. Single-Cell FISH and qPCR Validation Experiments
3. Results
4. Discussions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bushati, N.; Cohen, S.M. MicroRNA functions. Annu. Rev. Cell Dev. Biol. 2007, 23, 175–205. [Google Scholar] [CrossRef]
- Lu, J.; Guo, S.; Ebert, B.L.; Zhang, H.; Peng, X.; Bosco, J.; Pretz, J.; Schlanger, R.; Wang, J.Y.; Mak, R.H.; et al. MicroRNA-mediated control of cell fate in megakaryocyte-erythrocyte progenitors. Dev. Cell 2008, 14, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Sharp, P.A. GENE EXPRESSION Single-cell variability guided by microRNAs. Science 2016, 352, 1390–1391. [Google Scholar] [CrossRef] [PubMed]
- Denzler, R.; McGeary, S.E.; Title, A.C.; Agarwal, V.; Bartel, D.P.; Stoffel, M. Impact of MicroRNA Levels, Target-Site Complementarity, and Cooperativity on Competing Endogenous RNA-Regulated Gene Expression. Mol. Cell 2016, 64, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Schmiedel, J.M.; Klemm, S.L.; Zheng, Y.; Sahay, A.; Blüthgen, N.; Marks, D.S.; van Oudenaarden, A. MicroRNA control of protein expression noise. Science 2015, 348, 128–132. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef] [Green Version]
- Stelzer, Y.; Sagi, I.; Benvenisty, N. Involvement of parental imprinting in the antisense regulation of onco-miR-372-373. Nat. Commun. 2013, 4, 2724. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Wu, S.W.; Wu, W.P. A tumor-suppressive microRNA, miRNA-485-5p, inhibits glioma cell proliferation and invasion by down-regulating TPD52L2. Am. J. Transl. Res. 2017, 9, 3336–3344. [Google Scholar] [PubMed]
- Svoronos, A.A.; Engelman, D.M.; Slack, F.J. OncomiR or Tumor Suppressor? The Duplicity of MicroRNAs in Cancer. Cancer Res. 2016, 76, 3666–3670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levitin, H.M.; Yuan, J.; Sims, P.A. Single-Cell Transcriptomic Analysis of Tumor Heterogeneity. Trends Cancer 2018, 4, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Faridani, O.R.; Abdullayev, I.; Hagemann-Jensen, M.; Schell, J.P.; Lanner, F.; Sandberg, R. Single-cell sequencing of the small-RNA transcriptome. Nat. Biotechnol. 2016, 34, 1264–1266. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zheng, J.; Chen, Z.; Liu, Y.; Dura, B.; Kwak, M.; Xavier-Ferrucio, J.; Lu, Y.C.; Zhang, M.; Roden, C.; et al. Single-cell microRNA-mRNA co-sequencing reveals non-genetic heterogeneity and mechanisms of microRNA regulation. Nat. Commun. 2019, 10, 95. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Cheng, H.H.; Tewari, M. MicroRNA profiling: Approaches and considerations. Nat. Rev. Genet. 2012, 13, 358–369. [Google Scholar] [CrossRef]
- Roden, C.; Mastriano, S.; Wang, N.Y.; Lu, J. microRNA Expression Profiling: Technologies, Insights, and Prospects. Adv. Exp. Med. Biol. 2015, 888, 409–421. [Google Scholar]
- Li, J.; Smyth, P.; Cahill, S.; Denning, K.; Flavin, R.; Aherne, S.; Pirotta, M.; Guenther, S.M.; O’Leary, J.J.; Sheils, O. Improved RNA quality and TaqMan Pre-amplification method (PreAmp) to enhance expression analysis from formalin fixed paraffin embedded (FFPE) materials. BMC Biotechnol. 2008, 8, 10. [Google Scholar] [CrossRef]
- Ok, C.Y.; Singh, R.R.; Salim, A.A. Single-Cell Quantitative PCR: Advances and Potential in Cancer Diagnostics. Methods Mol. Biol. 2016, 1392, 153–159. [Google Scholar]
- Gong, H.; Do, D.; Ramakrishnan, R. Single-Cell mRNA-Seq Using the Fluidigm C1 System and Integrated Fluidics Circuits. Methods Mol. Biol. 2018, 1783, 193–207. [Google Scholar]
- Takousis, P. Analysis of Micro-RNA Expression by qPCR on a Microfluidics Platform for Alzheimer’s Disease Biomarker Discovery. Methods Mol. Biol. 2018, 1750, 283–292. [Google Scholar]
- Rossi, J.; Paczkowski, P.; Shen, Y.W.; Morse, K.; Flynn, B.; Kaiser, A.; Ng, C.; Gallatin, K.; Cain, T.; Fan, R.; et al. Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood 2018, 132, 804–814. [Google Scholar]
- Xue, Q.; Bettini, E.; Paczkowski, P.; Ng, C.; Kaiser, A.; McConnell, T.; Kodrasi, O.; Quigley, M.F.; Heath, J.; Fan, R.; et al. Single-cell multiplexed cytokine profiling of CD19 CAR-T cells reveals a diverse landscape of polyfunctional antigen-specific response. J. Immunother. Cancer 2017, 5, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Q.; Lu, Y.; Eisele, M.R.; Sulistijo, E.S.; Khan, N.; Fan, R.; Miller-Jensen, K. Analysis of single-cell cytokine secretion reveals a role for paracrine signaling in coordinating macrophage responses to TLR4 stimulation. Sci. Signal 2015, 8, ra59. [Google Scholar] [CrossRef]
- Lu, Y.; Xue, Q.; Eisele, M.R.; Sulistijo, E.S.; Brower, K.; Han, L.; Amir, E.A.D.; Pe’er, D.; Miller-Jensen, K.; Fan, R. Highly multiplexed profiling of single-cell effector functions reveals deep functional heterogeneity in response to pathogenic ligands. Proc. Natl. Acad. Sci. USA 2015, 112, E607–E615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. JAK-STAT Pathway Activation in Malignant and Nonmalignant Cells Contributes to MPN Pathogenesis and Therapeutic Response. Cancer Discov. 2015, 5, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chen, J.J.; Mu, L.; Xue, Q.; Wu, Y.; Wu, P.H.; Li, J.; Vortmeyer, A.O.; Miller-Jensen, K.; Wirtz, D.; et al. High-Throughput Secretomic Analysis of Single Cells to Assess Functional Cellular Heterogeneity. Anal. Chem. 2013, 85, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Whitesides, G.M.; Ostuni, E.; Takayama, S.; Jiang, X.; Ingber, D.E. Soft lithography in biology and biochemistry. Annu. Rev. Biomed. Eng. 2001, 3, 335–373. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Whitesides, G.M. Soft Lithography. Angew. Chem. Int. Ed. Engl. 1998, 37, 550–575. [Google Scholar] [CrossRef]
- Unger, M.A.; Chou, H.P.; Thorsen, T.; Scherer, A.; Quake, S.R. Monolithic microfabricated valves and pumps by multilayer soft lithography. Science 2000, 288, 113–116. [Google Scholar] [CrossRef]
- Giang, U.B.; Lee, D.; King, M.R.; DeLouise, L.A. Microfabrication of cavities in polydimethylsiloxane using DRIE silicon molds. Lab Chip 2007, 7, 1660–1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karadimitriou, N.K.; Joekar-Niasar, V.; Hassanizadeh, S.M.; Kleingeld, P.J.; Pyrak-Nolte, L.J. A novel deep reactive ion etched (DRIE) glass micro-model for two-phase flow experiments. Lab Chip 2012, 12, 3413–3418. [Google Scholar] [CrossRef]
- Shin, Y.S.; Ahmad, H.; Shi, Q.; Kim, H.; Pascal, T.A.; Fan, R.; Goddard, W.A., III; Heath, J.R. Chemistries for Patterning Robust DNA MicroBarcodes Enable Multiplex Assays of Cytoplasm Proteins from Single Cancer Cells. Chemphyschem 2010, 11, 3063–3069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Liu, C.; Chen, D.; Yang, W.H.; Liu, X.; Liu, C.G.; Dugas, C.M.; Tang, F.; Zheng, P.; Liu, Y.; et al. FOXP3 Controls an miR-146/NF-kappaB Negative Feedback Loop That Inhibits Apoptosis in Breast Cancer Cells. Cancer Res. 2015, 75, 1703–1713. [Google Scholar] [CrossRef]
- Guo, S.; Guo, W.; Li, S.; Dai, W.; Zhang, N.; Zhao, T.; Wang, H.; Ma, J.; Yi, X.; Ge, R.; et al. Serum miR-16: A Potential Biomarker for Predicting Melanoma Prognosis. J. Investig. Dermatol. 2016, 136, 985–993. [Google Scholar] [CrossRef]
- Pfeffer, S.R.; Yang, C.H.; Pfeffer, L.M. The Role of miR-21 in Cancer. Drug Dev. Res. 2015, 76, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Stinson, S.; Lackner, M.R.; Adai, A.T.; Yu, N.; Kim, H.J.; O’Brien, C.; Spoerke, J.; Jhunjhunwala, S.; Boyd, Z.; Januario, T.; et al. TRPS1 targeting by miR-221/222 promotes the epithelial-to-mesenchymal transition in breast cancer. Sci. Signal 2011, 4, ra41. [Google Scholar]
- Le Sage, C.; Nagel, R.; Egan, D.A.; Schrier, M.; Mesman, E.; Mangiola, A.; Anile, C.; Maira, G.; Mercatelli, N.; Ciafrè, S.A.; et al. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. Embo J. 2007, 26, 3699–3708. [Google Scholar] [CrossRef]
- Elitas, M.; Brower, K.; Lu, Y.; Chen, J.J.; Fan, R. A microchip platform for interrogating tumor-macrophage paracrine signaling at the single-cell level. Lab Chip 2014, 14, 3582–3588. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Piccini, M.; Koh, C.Y.; Lam, K.S.; Singh, A.K. Single cell microRNA analysis using microfluidic flow cytometry. PLoS ONE 2013, 8, e55044. [Google Scholar] [CrossRef]
- Lu, J.; Tsourkas, A. Imaging individual microRNAs in single mammalian cells in situ. Nucleic Acids Res. 2009, 37, e100. [Google Scholar] [CrossRef] [PubMed]
- Smibert, P.; Yang, J.S.; Azzam, G.; Liu, J.L.; Lai, E.C. Homeostatic control of Argonaute stability by microRNA availability. Nat. Struct. Mol. Biol. 2013, 20, 789–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golden, R.J.; Chen, B.; Li, T.; Braun, J.; Manjunath, H.; Chen, X.; Wu, J.; Schmid, V.; Chang, T.C.; Kopp, F.; et al. An Argonaute phosphorylation cycle promotes microRNA-mediated silencing. Nature 2017, 542, 197–202. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Niu, D.; Carbonell, A.; Wang, A.; Lee, A.; Tun, V.; Wang, Z.; Carrington, J.C.; Chia-en, A.C.; Jin, H. ARGONAUTE PIWI domain and microRNA duplex structure regulate small RNA sorting in Arabidopsis. Nat. Commun. 2014, 5, 5468. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.S.; Smibert, P.; Westholm, J.O.; Jee, D.; Maurin, T.; Lai, E.C. Intertwined pathways for Argonaute-mediated microRNA biogenesis in Drosophila. Nucleic Acids Res. 2014, 42, 1987–2002. [Google Scholar] [CrossRef] [PubMed]
- Hagemann-Jensen, M.; Abdullayev, I.; Sandberg, R.; Faridani, O.R. Small-seq for single-cell small-RNA sequencing. Nat. Protoc. 2018, 13, 2407–2424. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, N.; Lu, Y.; Chen, Z.; Fan, R. Multiplexed PCR-Free Detection of MicroRNAs in Single Cancer Cells Using a DNA-Barcoded Microtrough Array Chip. Micromachines 2019, 10, 215. https://doi.org/10.3390/mi10040215
Wang N, Lu Y, Chen Z, Fan R. Multiplexed PCR-Free Detection of MicroRNAs in Single Cancer Cells Using a DNA-Barcoded Microtrough Array Chip. Micromachines. 2019; 10(4):215. https://doi.org/10.3390/mi10040215
Chicago/Turabian StyleWang, Nayi, Yao Lu, Zhuo Chen, and Rong Fan. 2019. "Multiplexed PCR-Free Detection of MicroRNAs in Single Cancer Cells Using a DNA-Barcoded Microtrough Array Chip" Micromachines 10, no. 4: 215. https://doi.org/10.3390/mi10040215
APA StyleWang, N., Lu, Y., Chen, Z., & Fan, R. (2019). Multiplexed PCR-Free Detection of MicroRNAs in Single Cancer Cells Using a DNA-Barcoded Microtrough Array Chip. Micromachines, 10(4), 215. https://doi.org/10.3390/mi10040215