
A Microfluidic-Based Cell-Stretching Culture Device That Allows for Easy Preparation of Slides for Observation with High-Magnification Objective Lenses


Abstract
:1. Introduction
2. Materials and Methods
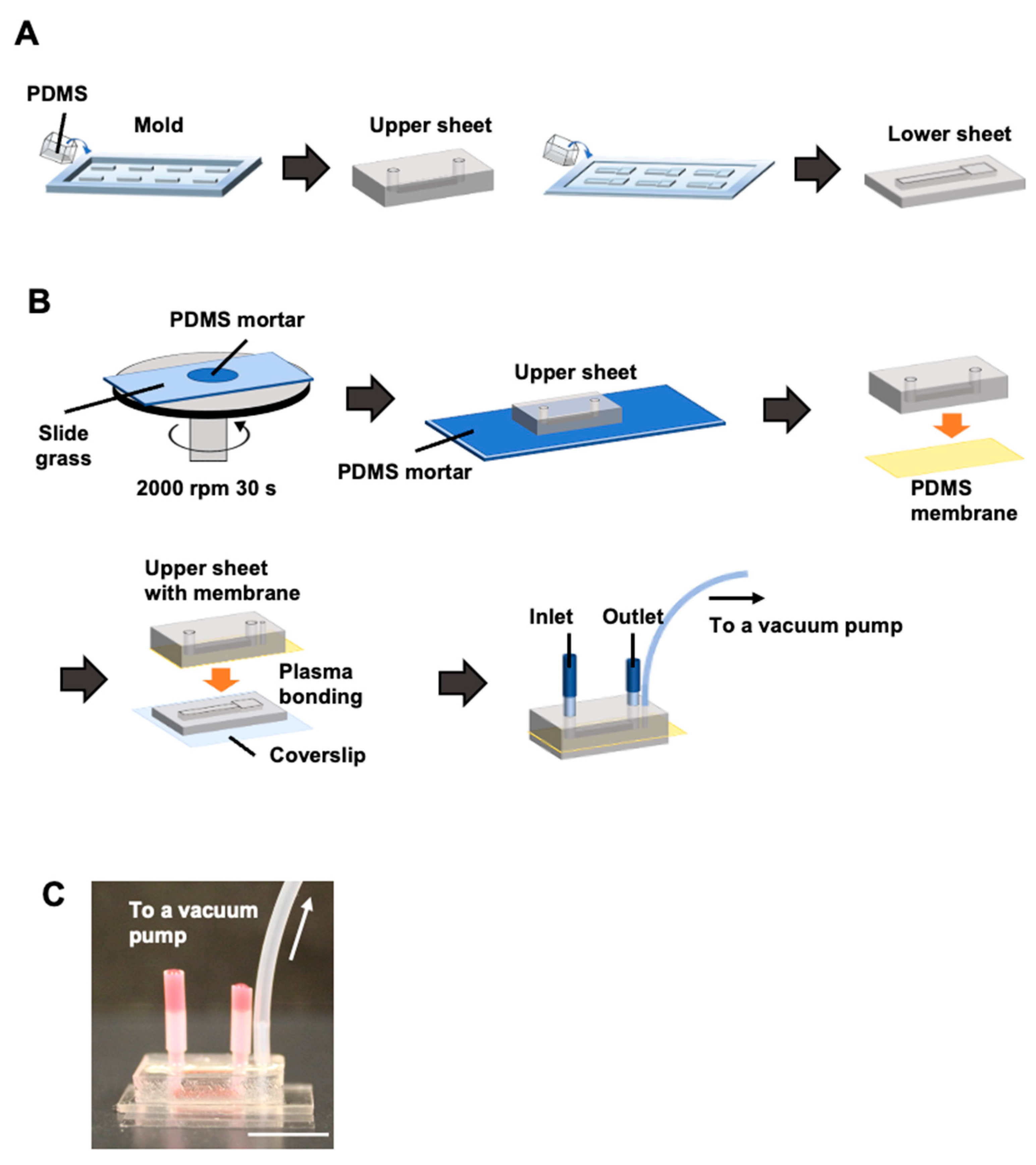
2.1. Fabrication of a Cell-Stretching Device
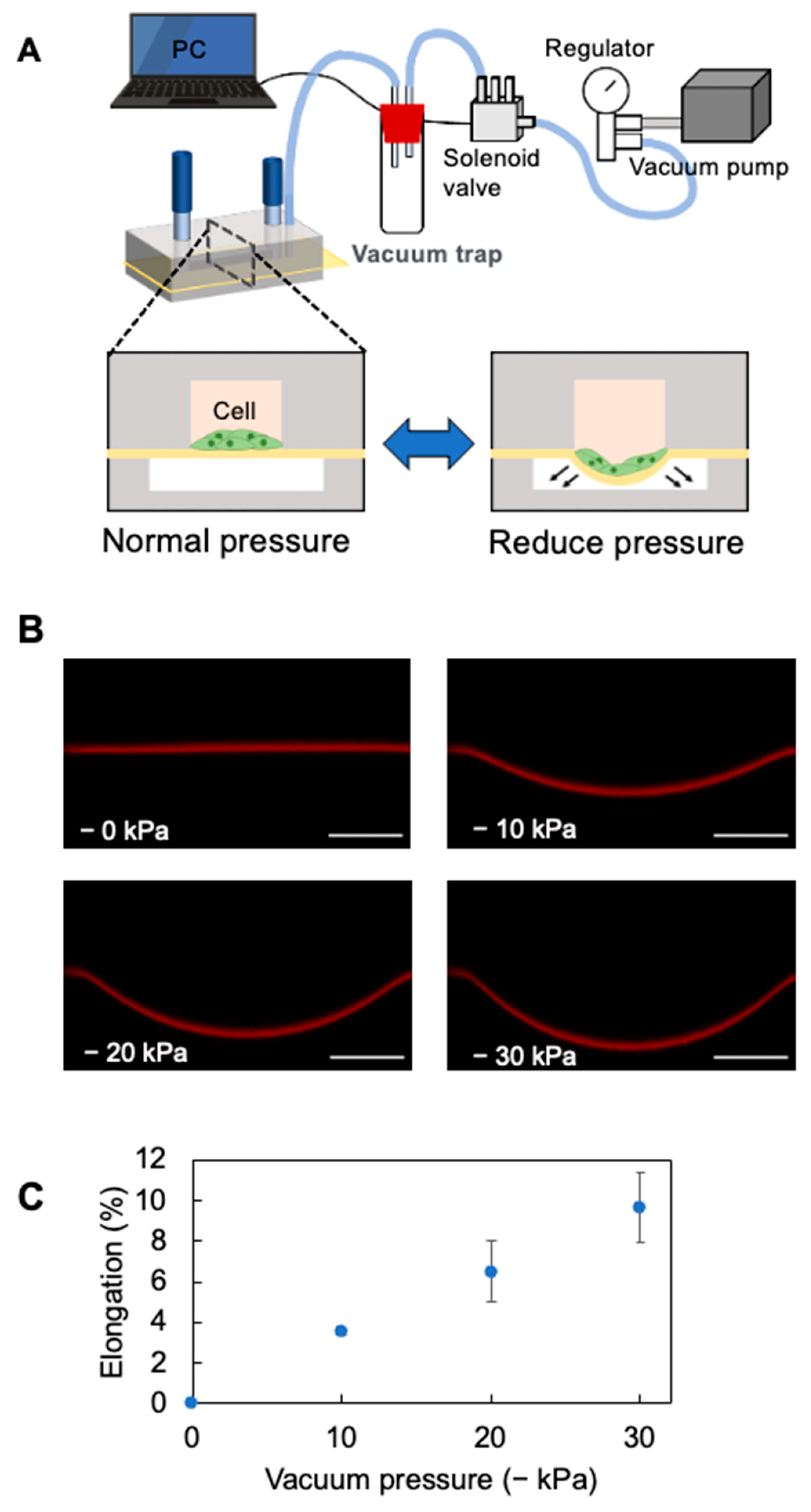
2.2. Cell Stretch System
2.3. Analysis of the Stretching Properties of the PDMS Membrane
2.4. Cell Culture in Cell-Stretching Devices
2.5. Cell Staining
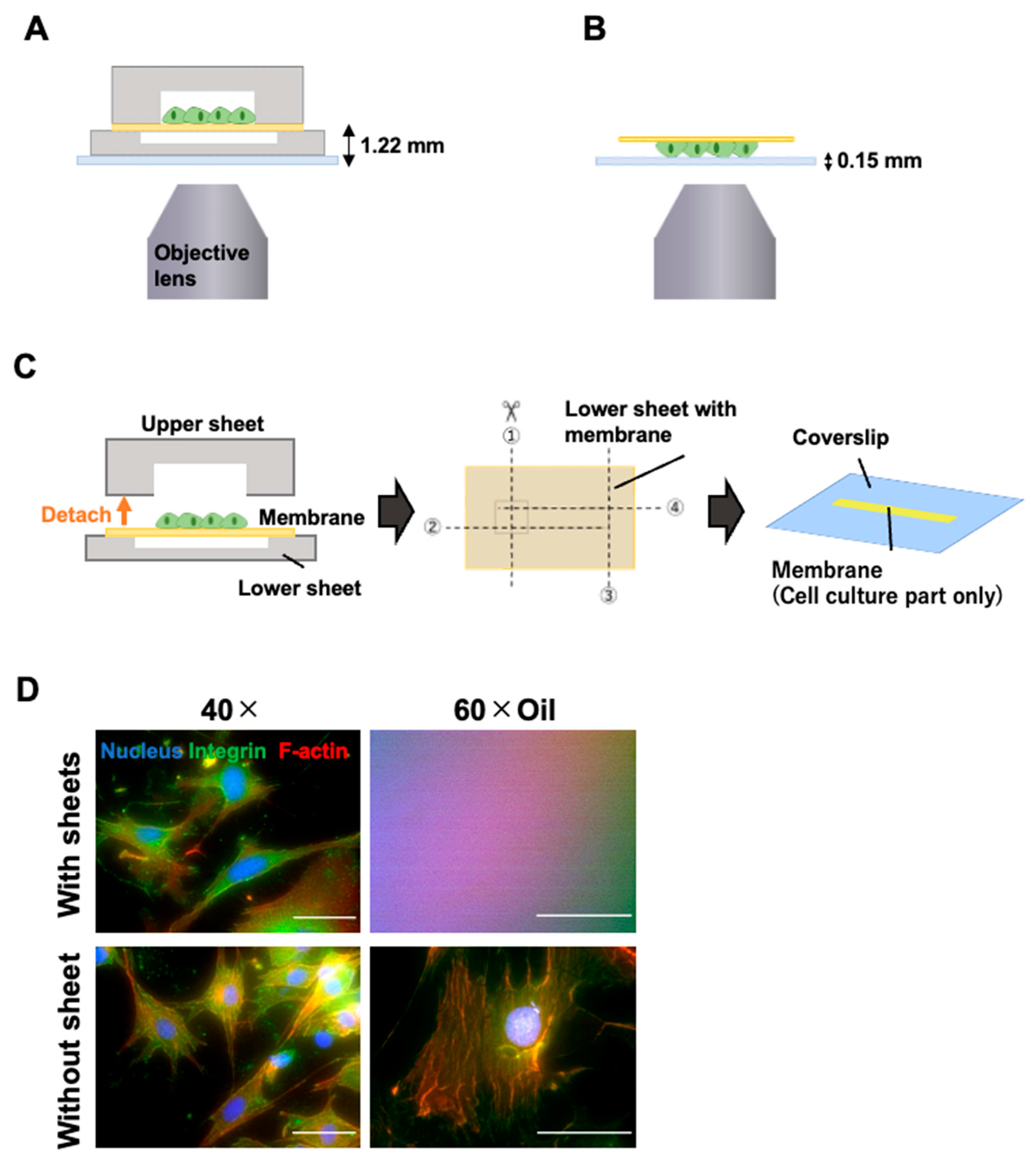
2.6. Microscopic Observation
3. Results and Discussion
3.1. Elongation Rate of the PDMS Thin Membrane in the Cell-Stretching Device
3.2. Microscopic Observation with High-Magnification Objective Lens
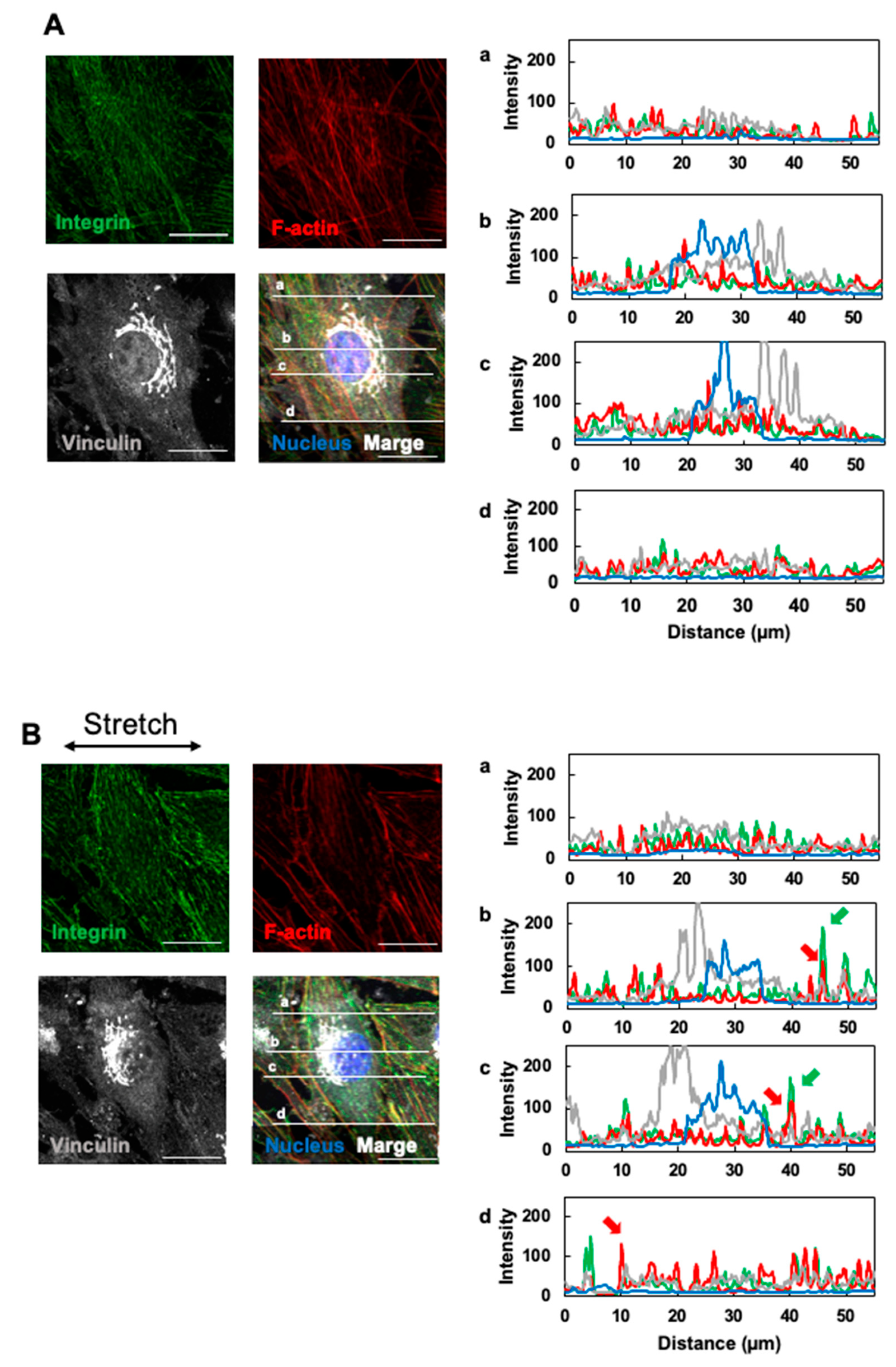
3.3. Effects of Cyclic Stretching on Stress Fibers and Focal Adhesion
3.4. Effects of Cyclic Stretching on YAP1 Intracellular Localization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cui, Y.; Hameed, F.M.; Yang, B.; Lee, K.; Pan, C.Q.; Park, S.; Sheetz, M. Cyclic stretching of soft substrates induces spreading and growth. Nat. Commun. 2015, 6, 6333. [Google Scholar] [CrossRef] [PubMed]
- Ladoux, B.; Mège, R.M. Mechanobiology of collective cell behaviours. Nat. Rev. Mol. Cell Biol. 2017, 18, 743–757. [Google Scholar] [CrossRef]
- Du, H.; Bartleson, J.M.; Butenko, S.; Alonso, V.; Liu, W.F.; Winer, D.A.; Butte, M.J. Tuning immunity through tissue mechanotransduction. Nat. Rev. Immunol. 2023, 23, 174–188. [Google Scholar] [CrossRef]
- Constantinou, I.; Bastounis, E.E. Cell-stretching devices: Advances and challenges in biomedical research and live-cell imaging. Trends Biotechnol. 2023, 41, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Polacheck, W.J.; Li, R.; Uzel, S.G.; Kamm, R.D. Microfluidic platforms for mechanobiology. Lab Chip 2013, 13, 2252–2267. [Google Scholar] [CrossRef] [PubMed]
- Kamble, H.; Barton, M.J.; Jun, M.; Park, S.; Nguyen, N.T. Cell stretching devices as research tools: Engineering and biological considerations. Lab Chip 2016, 16, 3193–3203. [Google Scholar] [CrossRef] [PubMed]
- Ribas, J.; Zhang, Y.S.; Pitrez, P.R.; Leijten, J.; Miscuglio, M.; Rouwkema, J.; Dokmeci, M.R.; Nissan, X.; Ferreira, L.; Khademhosseini, A. Biomechanical Strain Exacerbates Inflammation on a Progeria-on-a-Chip Model. Small 2017, 13, 1603737. [Google Scholar] [CrossRef] [PubMed]
- Kaarj, K.; Yoon, J.Y. Methods of Delivering Mechanical Stimuli to Organ-on-a-Chip. Micromachines 2019, 10, 700. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Nitta, M.; Ogawa, A. A microfluidic cell stretch device to investigate the effects of stretching stress on artery smooth muscle cell proliferation in pulmonary arterial hypertension. Inventions 2019, 4, 1. [Google Scholar] [CrossRef]
- Thompson, C.L.; Fu, S.; Knight, M.M.; Thorpe, S.D. Mechanical Stimulation: A Crucial Element of Organ-on-Chip Models. Front. Bioeng. Biotechnol. 2020, 8, 602646. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.; Thurgood, P.; Sekar, N.C.; Chen, S.; Pirogova, E.; Peter, K.; Baratchi, S.; Khoshmanesh, K. Microfluidic models of the human circulatory system: Versatile platforms for exploring mechanobiology and disease modeling. Biophys. Rev. 2021, 13, 769–786. [Google Scholar] [CrossRef] [PubMed]
- Onal, S.; Alkaisi, M.M.; Nock, V. Microdevice-based mechanical compression on living cells. iScience 2022, 25, 105518. [Google Scholar] [CrossRef] [PubMed]
- Jungbauer, S.; Gao, H.; Spatz, J.P.; Kemkemer, R. Two characteristic regimes in frequency-dependent dynamic reorientation of fibroblasts on cyclically stretched substrates. Biophys. J. 2008, 95, 3470–3478. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting organ-level lung functions on a chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Huh, D.; Hamilton, G.; Ingber, D.E. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip 2012, 12, 2165–2174. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.; Kim, H.J. 3D in vitro morphogenesis of human intestinal epithelium in a gut-on-a-chip or a hybrid chip with a cell culture insert. Nat. Protoc. 2022, 17, 910–939. [Google Scholar] [CrossRef]
- Valiei, A.; Aminian-Dehkordi, J.; Mofrad, M.R.K. Gut-on-a-chip models for dissecting the gut microbiology and physiology. APL Bioeng. 2023, 7, 011502. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.J.; Mehr, A.P.; Hamilton, G.A.; McPartlin, L.A.; Chung, S.; Suh, K.Y.; Ingber, D.E. Human kidney proximal tubule-on-a-chip for drug transport and nephrotoxicity assessment. Integr. Biol. 2013, 5, 1119–1129. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.J.; Otieno, M.A.; Ronxhi, J.; Lim, H.K.; Ewart, L.; Kodella, K.R.; Petropolis, D.B.; Kulkarni, G.; Rubins, J.E.; Conegliano, D.; et al. Reproducing human and cross-species drug toxicities using a Liver-Chip. Sci. Transl. Med. 2019, 11, eaax5516. [Google Scholar] [CrossRef] [PubMed]
- Grassart, A.; Malardé, V.; Gobaa, S.; Sartori-Rupp, A.; Kerns, J.; Karalis, K.; Marteyn, B.; Sansonetti, P.; Sauvonnet, N. Bioengineered Human Organ-on-Chip Reveals Intestinal Microenvironment and Mechanical Forces Impacting Shigella Infection. Cell Host Microbe 2019, 26, 435–444.e434. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, G.; Doherty, E.; To, T.; Sutherland, A.; Grant, J.; Junaid, A.; Gulati, A.; LoGrande, N.; Izadifar, Z.; Timilsina, S.S.; et al. Vaginal microbiome-host interactions modeled in a human vagina-on-a-chip. Microbiome 2022, 10, 201. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Mao, T.; Gu, Y.; Yang, Y.; Ding, J. A simplified yet enhanced and versatile microfluidic platform for cyclic cell stretching on an elastic polymer. Biofabrication 2020, 12, 045032. [Google Scholar] [CrossRef]
- Mao, T.; He, Y.; Gu, Y.; Yang, Y.; Yu, Y.; Wang, X.; Ding, J. Critical Frequency and Critical Stretching Rate for Reorientation of Cells on a Cyclically Stretched Polymer in a Microfluidic Chip. ACS Appl. Mater. Interfaces 2021, 13, 13934–13948. [Google Scholar] [CrossRef]
- Nakano, T.; Kodama, H.; Honjo, T. Generation of lymphohematopoietic cells from embryonic stem cells in culture. Science 1994, 265, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Lundin, V.; Sugden, W.W.; Theodore, L.N.; Sousa, P.M.; Han, A.; Chou, S.; Wrighton, P.J.; Cox, A.G.; Ingber, D.E.; Goessling, W.; et al. YAP Regulates Hematopoietic Stem Cell Formation in Response to the Biomechanical Forces of Blood Flow. Dev. Cell 2020, 52, 446–460.e445. [Google Scholar] [CrossRef]
- Sato, M.; Sasaki, N.; Ato, M.; Hirakawa, S.; Sato, K.; Sato, K. Microcirculation-on-a-Chip: A Microfluidic Platform for Assaying Blood- and Lymphatic-Vessel Permeability. PLoS ONE 2015, 10, e0137301. [Google Scholar] [CrossRef] [PubMed]
- Con, C.; Cui, B. Effect of mold treatment by solvent on PDMS molding into nanoholes. Nanoscale Res. Lett. 2013, 8, 394. [Google Scholar] [CrossRef]
- Litzenberger, J.B.; Kim, J.B.; Tummala, P.; Jacobs, C.R. Beta1 integrins mediate mechanosensitive signaling pathways in osteocytes. Calcif. Tissue Int. 2010, 86, 325–332. [Google Scholar] [CrossRef]
- Atherton, P.; Stutchbury, B.; Jethwa, D.; Ballestrem, C. Mechanosensitive components of integrin adhesions: Role of vinculin. Exp. Cell Res. 2016, 343, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Amiri, S.; Muresan, C.; Shang, X.; Huet-Calderwood, C.; Schwartz, M.A.; Calderwood, D.A.; Murrell, M. Intracellular tension sensor reveals mechanical anisotropy of the actin cytoskeleton. Nat. Commun. 2023, 14, 8011. [Google Scholar] [CrossRef]
- Henshaw, D.R.; Attia, E.; Bhargava, M.; Hannafin, J.A. Canine ACL fibroblast integrin expression and cell alignment in response to cyclic tensile strain in three-dimensional collagen gels. J. Orthop. Res. 2006, 24, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, D.; Sasazaki, Y.; Kikuchi, T.; Ono, T.; Nemoto, K.; Matsumoto, H.; Toyama, Y. Temporal effects of cyclic stretching on distribution and gene expression of integrin and cytoskeleton by ligament fibroblasts in vitro. Connect. Tissue Res. 2009, 50, 263–269. [Google Scholar] [CrossRef]
- Freeman, S.A.; Christian, S.; Austin, P.; Iu, I.; Graves, M.L.; Huang, L.; Tang, S.; Coombs, D.; Gold, M.R.; Roskelley, C.D. Applied stretch initiates directional invasion through the action of Rap1 GTPase as a tension sensor. J. Cell Sci. 2017, 130, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Albinsson, S.; Nordström, I.; Hellstrand, P. Stretch of the vascular wall induces smooth muscle differentiation by promoting actin polymerization. J. Biol. Chem. 2004, 279, 34849–34855. [Google Scholar] [CrossRef] [PubMed]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Qu, R.; Feng, Y.; Huang, X.; Yang, Y.; Fan, T.; Sun, B.; Khan, A.U.; Wu, S.; Dai, J.; et al. Regulation of the integrin αVβ3- actin filaments axis in early osteogenesis of human fibroblasts under cyclic tensile stress. Stem Cell. Res. Ther. 2021, 12, 523. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Riehl, B.D.; Bouzid, T.; Yang, R.; Duan, B.; Donahue, H.J.; Lim, J.Y. YAP mechanotransduction under cyclic mechanical stretch loading for mesenchymal stem cell osteogenesis is regulated by ROCK. Front. Bioeng. Biotechnol. 2024, 11, 1306002. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kato, M.; Sato, K. A Microfluidic-Based Cell-Stretching Culture Device That Allows for Easy Preparation of Slides for Observation with High-Magnification Objective Lenses. Micromachines 2025, 16, 93. https://doi.org/10.3390/mi16010093
Kato M, Sato K. A Microfluidic-Based Cell-Stretching Culture Device That Allows for Easy Preparation of Slides for Observation with High-Magnification Objective Lenses. Micromachines. 2025; 16(1):93. https://doi.org/10.3390/mi16010093
Chicago/Turabian StyleKato, Momoko, and Kae Sato. 2025. "A Microfluidic-Based Cell-Stretching Culture Device That Allows for Easy Preparation of Slides for Observation with High-Magnification Objective Lenses" Micromachines 16, no. 1: 93. https://doi.org/10.3390/mi16010093
APA StyleKato, M., & Sato, K. (2025). A Microfluidic-Based Cell-Stretching Culture Device That Allows for Easy Preparation of Slides for Observation with High-Magnification Objective Lenses. Micromachines, 16(1), 93. https://doi.org/10.3390/mi16010093