
Microfabricated Physiological Models for In Vitro Drug Screening Applications

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
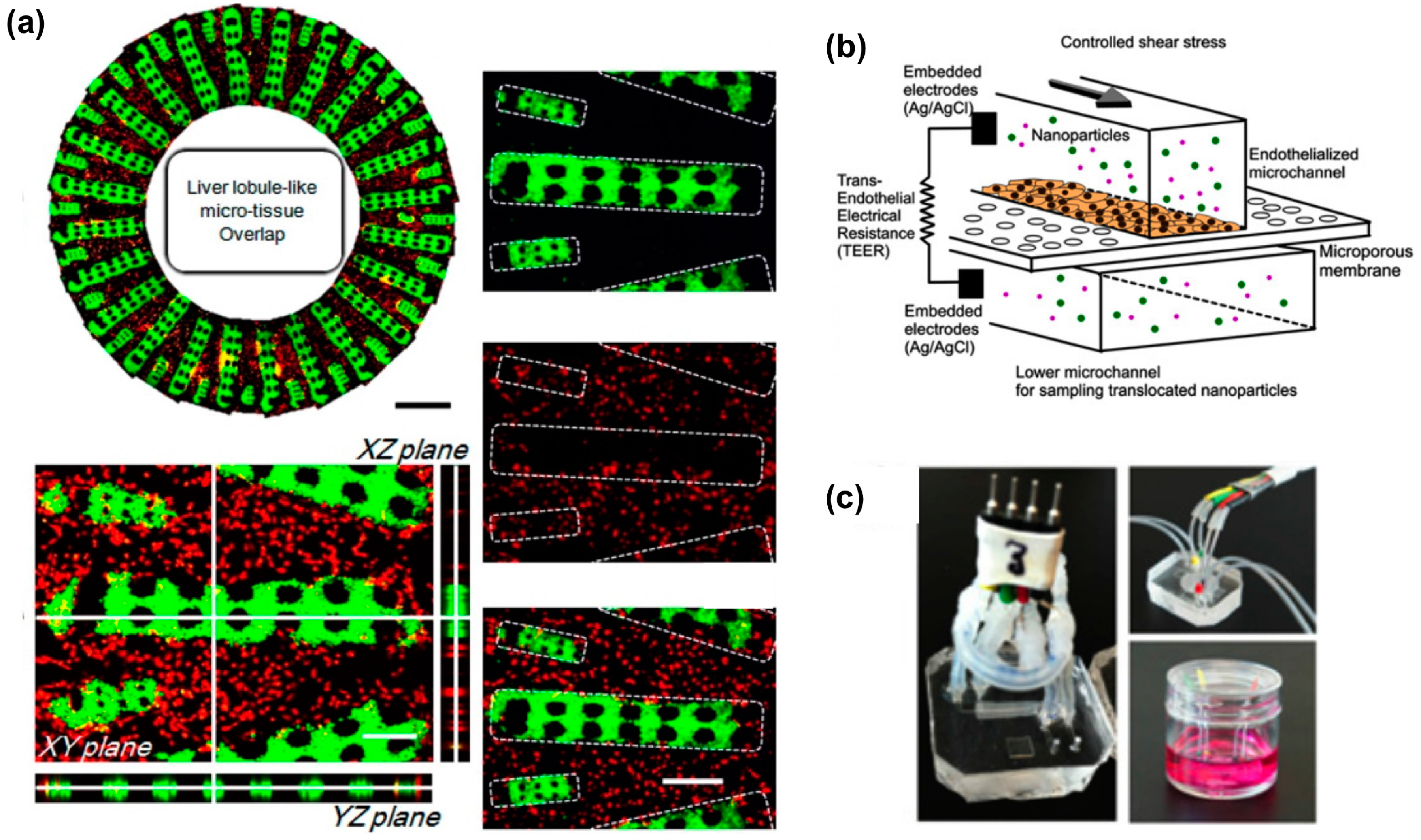
2. Liver
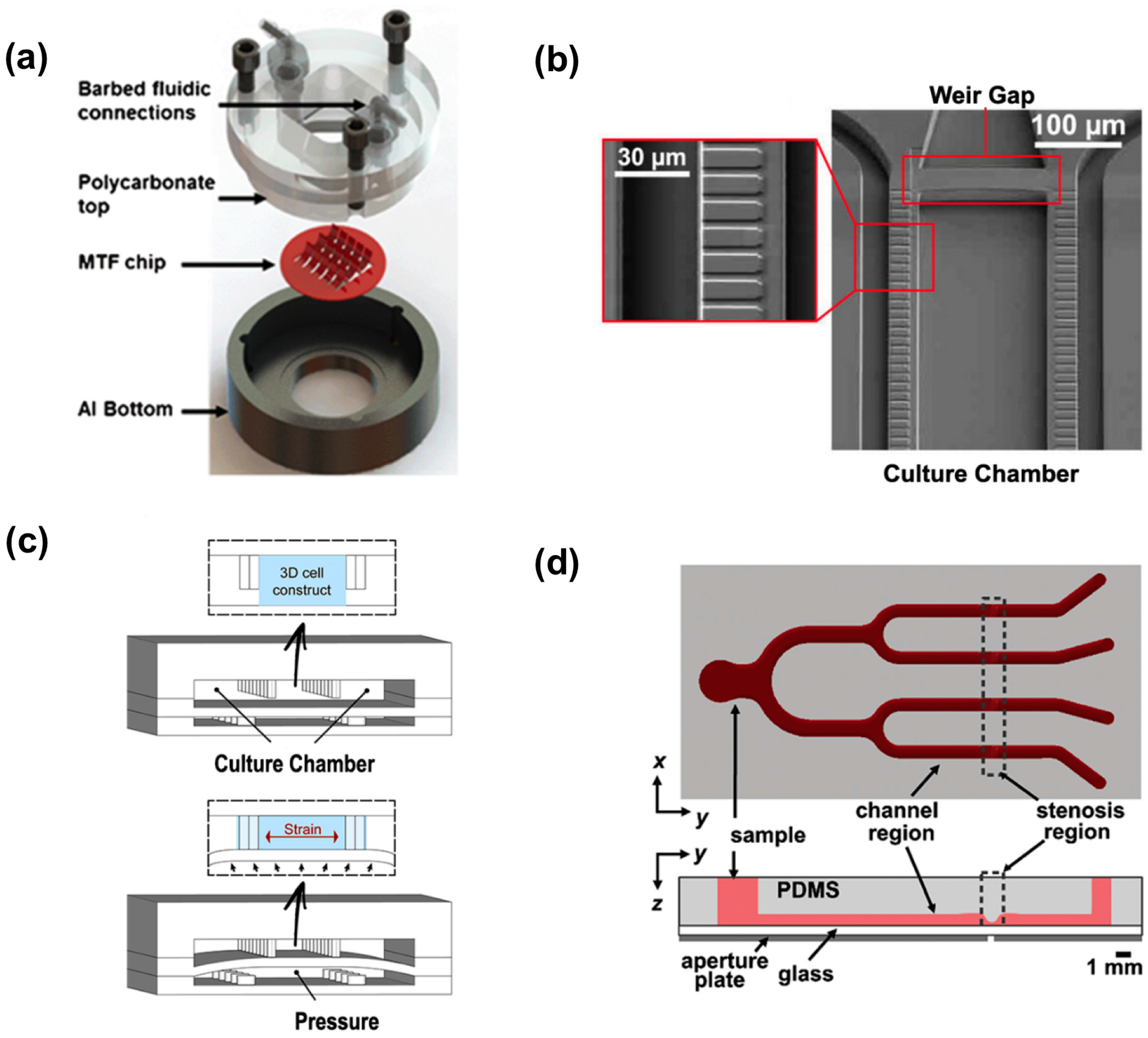
3. Central Nervous System
4. Heart
5. Vascular System
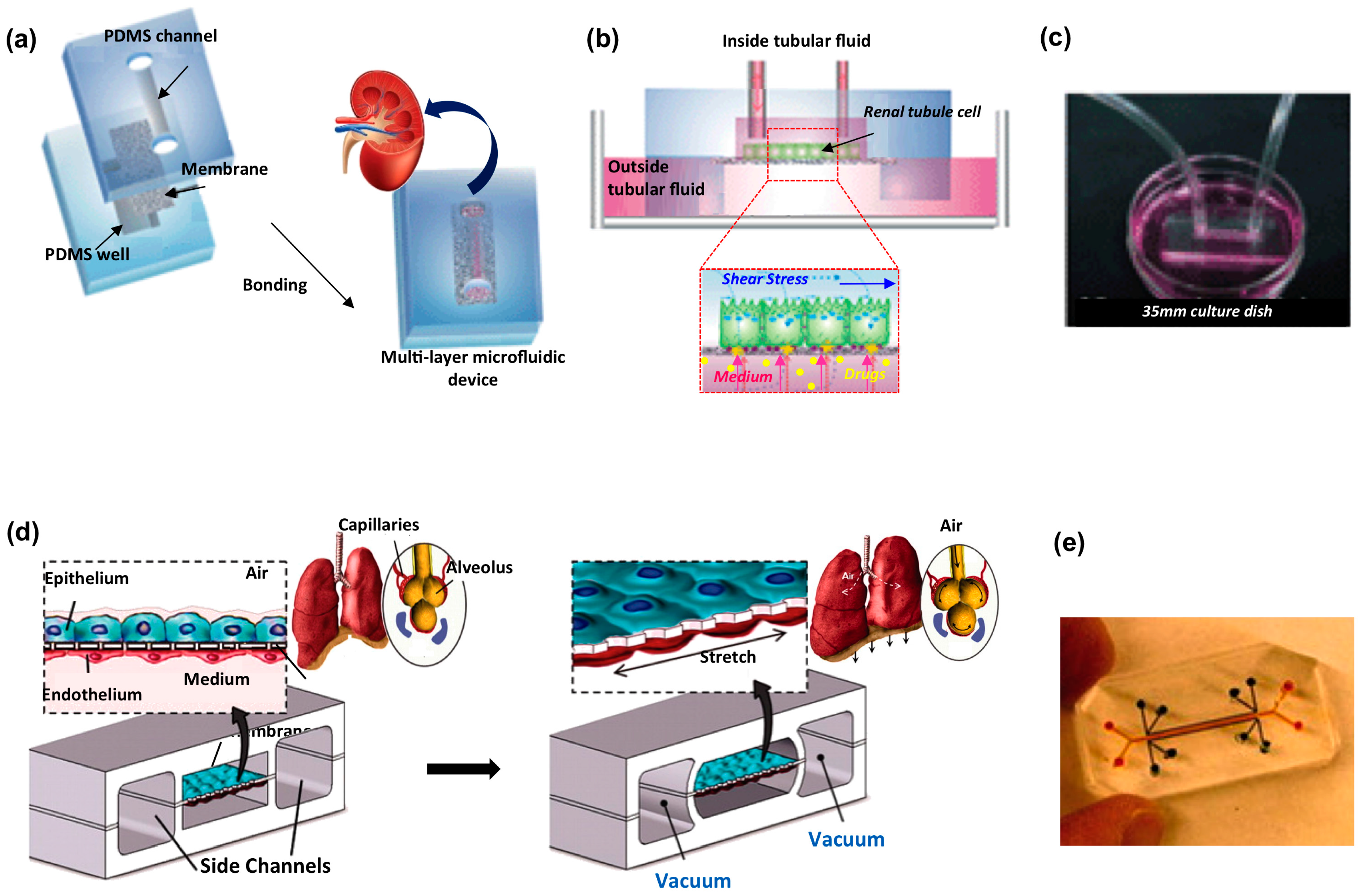
6. Lung
7. Kidney
8. Perspective Outlook: Body-on-Chips
Acknowledgments
Author Contributions
Conflicts of Interest
References
- DiMasi, J.A.; Hansen, R.W.; Grabowski, H.G. The price of innovation: New estimates of drug development costs. J. Health Econ. 2003, 22, 151–185. [Google Scholar] [CrossRef]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [PubMed]
- Lipsky, M.S.; Sharp, L.K. From idea to market: The drug approval process FDA: A historical perspective. Development 2001, 14, 362–367. [Google Scholar]
- Costa, A.; Sarmento, B.; Seabra, V. An evaluation of the latest in vitro tools for drug metabolism studies. Expert Opin. Drug Metab. Toxicol 2014, 10, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Wienkers, L.C.; Heath, T.G. Predicting in vivo drug interactions from in vitro drug discovery data. Nat. Rev. Drug Discov. 2005, 4, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Cook, N.; Jodrell, D.I.; Tuveson, D.A. Predictive in vivo animal models and translation to clinical trials. Drug Discov. Today 2012, 17, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Pammolli, F.; Magazzini, L.; Riccaboni, M. The productivity crisis in pharmaceutical R&D. Nat. Rev. Drug Discov. 2011, 10, 428–438. [Google Scholar] [PubMed]
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 1–5. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. The cost of drug development. N. Engl. J. Med. 2015, 372, 1972. [Google Scholar] [CrossRef] [PubMed]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar] [PubMed]
- Schuh, J.C.L. Trials, tribulations, and trends in tumor modeling in mice. Toxicol. Pathol. 2004, 32 (Suppl. 1), 53–66. [Google Scholar] [CrossRef] [PubMed]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef] [PubMed]
- Whitesides, G.M. The origins and the future of microfluidics. Nature 2006, 442, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Sackmann, E.K.; Fulton, A.L.; Beebe, D.J. The present and future role of microfluidics in biomedical research. Nature 2014, 507, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Hamilton, G.A.; Ingber, D.E. From 3D cell culture to organs-on-chips. Trends Cell Biol. 2011, 21, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Torisawa, Y.; Hamilton, G.A.; Kim, H.J.; Ingber, D.E. Microengineered physiological biomimicry: Organs-on-Chips. Lab Chip 2012, 12, 2156–2164. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Yum, K.; Hong, S.G.; Healy, K.E.; Lee, L.P. Physiologically relevant organs on chips. Biotechnol. J. 2014, 9, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Fu, F.; Cheng, Y.; Wang, C.; Zhao, Y.; Gu, Z. Organ-on-a-Chip Systems: Microengineering to biomimic living systems. Small 2016, 12, 2253–2282. [Google Scholar] [CrossRef] [PubMed]
- Perestrelo, A.R.; Águas, A.C.P.; Rainer, A.; Forte, G. Microfluidic organ/body-on-a-chip devices at the convergence of biology and microengineering. Sensors 2015, 15, 29848. [Google Scholar] [CrossRef] [PubMed]
- Li, A.P.; Lu, C.; Brent, J.A.; Pham, C.; Fackett, A.; Ruegg, C.E.; Silber, P.M. Cryopreserved human hepatocytes: Characterization of drug-metabolizing activities and applications in higher throughput screening assays for hepatotoxicity, metabolic stability, and drug-drug interaction potential. Chem. Biol. Interact. 1999, 121, 17–35. [Google Scholar] [CrossRef]
- Wrighton, S.A.; Stevens, J.C. The human hepatic cytochromes P450 involved in drug metabolism. Crit. Rev. Toxicol. 1992, 22, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Gerets, H.H.J.; Tilmant, K.; Gerin, B.; Chanteux, H.; Depelchin, B.O.; Dhalluin, S.; Atienzar, F.A. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biol. Toxicol. 2012, 28, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Ravi, M.; Paramesh, V.; Kaviya, S.R.; Anuradha, E.; Paul Solomon, F.D. 3D cell culture systems: Advantages and applications. J. Cell. Physiol. 2015, 230, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Tibbitt, M.W.; Anseth, K.S. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng. 2009, 103, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Van Duinen, V.; Trietsch, S.J.; Joore, J.; Vulto, P.; Hankemeier, T. Microfluidic 3D cell culture: From tools to tissue models. Curr. Opin. Biotechnol. 2015, 35, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.Y.; Zhou, L.H.; Zhang, Q.C.; Chen, Y.M.; Sun, W.; Xu, F.; Lu, T.J. Microfluidic hydrogels for tissue engineering. Biofabrication 2011, 3, 12001. [Google Scholar] [CrossRef] [PubMed]
- Toh, Y.-C.; Zhang, C.; Zhang, J.; Khong, Y.M.; Chang, S.; Samper, V.D.; van Noort, D.; Hutmacher, D.W.; Yu, H. A novel 3D mammalian cell perfusion-culture system in microfluidic channels. Lab Chip 2007, 7, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Au, S.H.; Chamberlain, M.D.; Mahesh, S.; Sefton, M.V.; Wheeler, A.R. Hepatic organoids for microfluidic drug screening. Lab Chip 2014, 14, 3290–3299. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chia, S.M.; Ong, S.M.; Zhang, S.; Toh, Y.C.; van Noort, D.; Yu, H. The controlled presentation of TGF-beta1 to hepatocytes in a 3D-microfluidic cell culture system. Biomaterials 2009, 30, 3847–3853. [Google Scholar] [CrossRef] [PubMed]
- Toh, Y.-C.; Lim, T.C.; Tai, D.; Xiao, G.; van Noort, D.; Yu, H. A microfluidic 3D hepatocyte chip for drug toxicity testing. Lab Chip 2009, 9, 2026–2035. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Yeon, J.H.; Park, J.-K. A microfluidic platform for 3-dimensional cell culture and cell-based assays. Biomed. Microdevices 2007, 9, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Bhise, N.S.; Manoharan, V.; Massa, S.; Tamayol, A.; Ghaderi, M.; Miscuglio, M.; Lang, Q.; Zhang, Y.S.; Shin, S.R.; Calzone, G.; et al. A liver-on-a-chip platform with bioprinted hepatic spheroids. Biofabrication 2016, 8, 14101. [Google Scholar] [CrossRef] [PubMed]
- Dash, A.; Inman, W.; Hoffmaster, K.; Sevidal, S.; Kelly, J.; Obach, R.S.; Griffith, L.G.; Tannenbaum, S.R. Liver tissue engineering in the evaluation of drug safety. Expert Opin. Drug Metab. Toxicol. 2009, 5, 1159–1174. [Google Scholar] [CrossRef] [PubMed]
- Soldatow, V.Y.; LeCluyse, E.L.; Griffith, L.G.; Rusyn, I. In vitro models for liver toxicity testing. Toxicol. Res. (Camb.) 2013, 2, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.B.A.; Rawat, S.; Cirillo, J.; Bouchard, M.; Noh, H.M. Layered long-term co-culture of hepatocytes and endothelial cells on a transwell membrane: Toward engineering the liver sinusoid. Biofabrication 2013, 5, 45008. [Google Scholar] [CrossRef] [PubMed]
- Ohno, M.; Motojima, K.; Okano, T.; Taniguchi, A. Up-regulation of drug-metabolizing enzyme genes in layered co-culture of a human liver cell line and endothelial cells. Tissue Eng. Part A 2008, 14, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, A.; Senutovitch, N.; Bale, S.S.; McCarty, W.J.; Hegde, M.; Jindal, R.; Golberg, I.; Berk Usta, O.; Yarmush, M.L.; Vernetti, L.; et al. Towards a three-dimensional microfluidic liver platform for predicting drug efficacy and toxicity in humans. Stem Cell Res. Ther. 2013, 4 (Suppl. 1), S16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Patel, D.; Kwa, T.; Haque, A.; Matharu, Z.; Stybayeva, G.; Gao, Y.; Diehl, A.M.; Revzin, A. Liver injury-on-a-chip: Microfluidic co-cultures with integrated biosensors for monitoring liver cell signaling during injury. Lab Chip 2015, 15, 4467–4478. [Google Scholar] [CrossRef] [PubMed]
- Rennert, K.; Steinborn, S.; Gröger, M.; Ungerböck, B.; Jank, A.M.; Ehgartner, J.; Nietzsche, S.; Dinger, J.; Kiehntopf, M.; Funke, H.; et al. A microfluidically perfused three dimensional human liver model. Biomaterials 2015, 71, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Khetani, S.R.; Bhatia, S.N. Microscale culture of human liver cells for drug development. Nat. Biotechnol. 2008, 26, 120–126. [Google Scholar] [CrossRef] [PubMed]
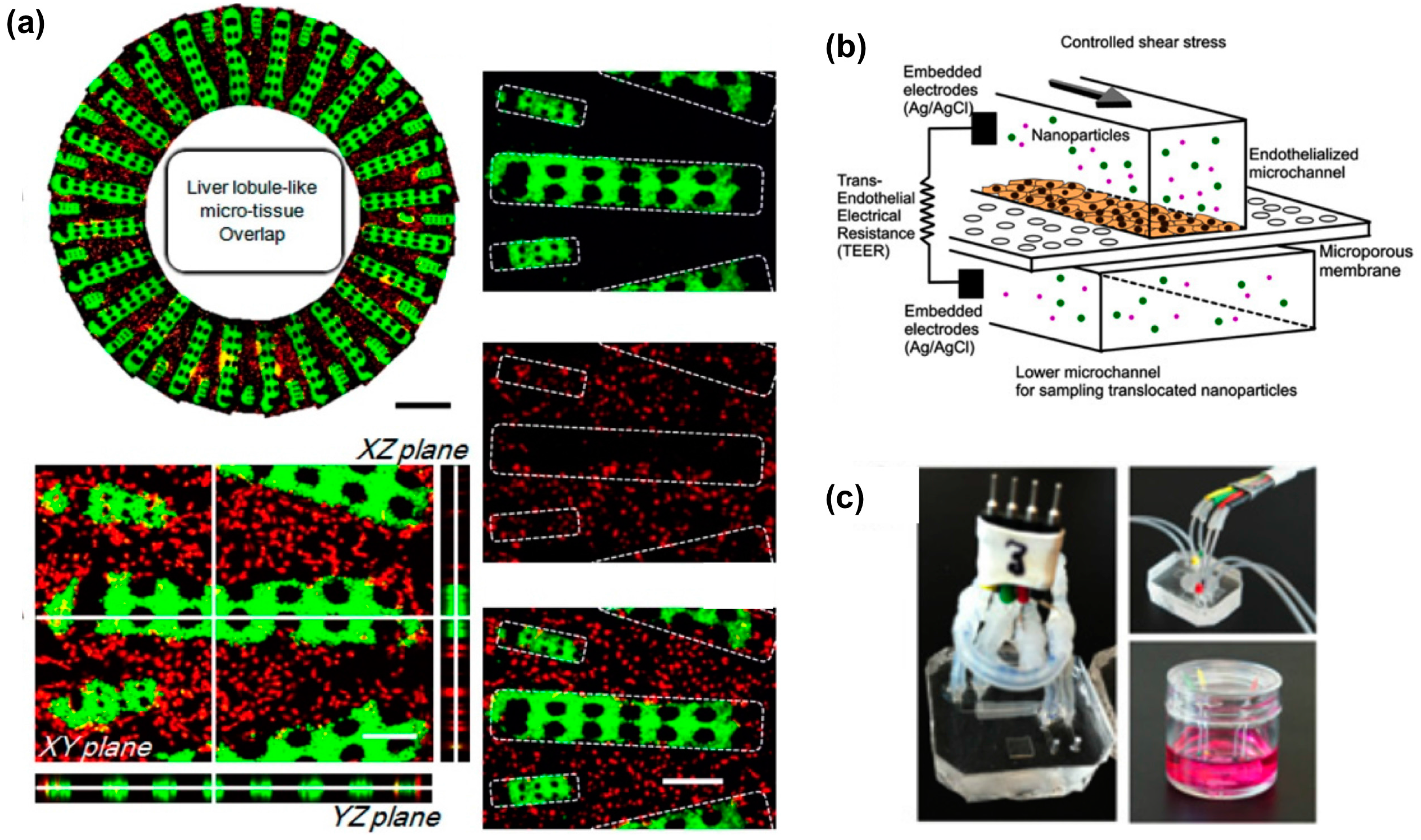
- Ma, C.; Zhao, L.; Zhou, E.M.; Xu, J.; Shen, S.; Wang, J. On-chip construction of liver lobule-like microtissue and its application for adverse drug reaction assay. Anal. Chem. 2016, 88, 1719–1727. [Google Scholar] [CrossRef] [PubMed]
- Prot, J.M.; Aninat, C.; Griscom, L.; Razan, F.; Brochot, C.; Guillouzo, C.G.; Legallais, C.; Corlu, A.; Leclerc, E. Improvement of HepG2/C3a cell functions in a microfluidic biochip. Biotechnol. Bioeng. 2011, 108, 1704–1715. [Google Scholar] [CrossRef] [PubMed]
- Baudoin, R.; Legendre, A.; Jacques, S.; Cotton, J.; Bois, F.; Leclerc, E. Evaluation of a liver microfluidic biochip to predict in vivo clearances of seven drugs in rats. J. Pharm. Sci. 2014, 103, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Prot, J.M.; Briffaut, A.S.; Letourneur, F.; Chafey, P.; Merlier, F.; Grandvalet, Y.; Legallais, C.; Leclerc, E. Integrated proteomic and transcriptomic investigation of the acetaminophen toxicity in liver microfluidic biochip. PLoS ONE 2011, 6, e21268. [Google Scholar] [CrossRef] [PubMed]
- Prot, J.M.; Videau, O.; Brochot, C.; Legallais, C.; Bénech, H.; Leclerc, E. A cocktail of metabolic probes demonstrates the relevance of primary human hepatocyte cultures in a microfluidic biochip for pharmaceutical drug screening. Int. J. Pharm. 2011, 408, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Baudoin, R.; Griscom, L.; Prot, J.M.; Legallais, C.; Leclerc, E. Behavior of HepG2/C3A cell cultures in a microfluidic bioreactor. Biochem. Eng. J. 2011, 53, 172–181. [Google Scholar] [CrossRef]
- Lee, P.J.; Hung, P.J.; Lee, L.P. An artificial liver sinusoid with a microfluidic endothelial-like barrier for primary hepatocyte culture. Biotechnol. Bioeng. 2007, 97, 1340–1346. [Google Scholar] [CrossRef] [PubMed]
- Esch, M.B.; Prot, J.-M.; Wang, Y.I.; Miller, P.; Llamas-Vidales, J.R.; Naughton, B.A.; Applegate, D.R.; Shuler, M.L.; Facility, C.N. Multi-cellular 3D human primary liver cell culture elevates metabolic activity under fluidic flow. Lab Chip 2015, 15, 2269–2277. [Google Scholar] [CrossRef] [PubMed]
- Carraro, A.; Hsu, W.M.; Kulig, K.M.; Cheung, W.S.; Miller, M.L.; Weinberg, E.J.; Swart, E.F.; Kaazempur-Mofrad, M.; Borenstein, J.T.; Vacanti, J.P.; et al. In vitro analysis of a hepatic device with intrinsic microvascular-based channels. Biomed. Microdevices 2008, 10, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Novik, E.; Maguire, T.J.; Chao, P.; Cheng, K.C.; Yarmush, M.L. A microfluidic hepatic coculture platform for cell-based drug metabolism studies. Biochem. Pharmacol. 2010, 79, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lobatto, M.E.; Kawahara, T.; Lee Chung, B.; Mieszawska, A.J.; Sanchez-Gaytan, B.L.; Fay, F.; Senders, M.L.; Calcagno, C.; Becraft, J.; et al. Probing nanoparticle translocation across the permeable endothelium in experimental atherosclerosis. Proc. Natl. Acad. Sci. USA 2014, 111, 1078–1083. [Google Scholar] [CrossRef] [PubMed]
- DiNunzio, J.C.; Williams, R.O. CNS disorders--current treatment options and the prospects for advanced therapies. Drug Dev. Ind. Pharm. 2008, 34, 1141–1167. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.G.; Flanagan, L.A.; Rhee, S.W.; Schwartz, P.H.; Lee, A.P.; Monuki, E.S.; Jeon, N.L. Human neural stem cell growth and differentiation in a gradient-generating microfluidic device. Lab Chip 2005, 5, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Kapur, T.A.; Shoichet, M.S. Immobilized concentration gradients of nerve growth factor guide neurite outgrowth. J. Biomed. Mater. Res. A 2004, 68, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Romano, N.H.; Lampe, K.J.; Xu, H.; Ferreira, M.M.; Heilshorn, S.C. Microfluidic gradients reveal enhanced neurite outgrowth but impaired guidance within 3D matrices with high integrin ligand densities. Small 2015, 11, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Kunze, A.; Meissner, R.; Brando, S.; Renaud, P. Co-pathological connected primary neurons in a microfluidic device for Alzheimer studies. Biotechnol. Bioeng. 2011, 108, 2241–2245. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.R.; Zeng, W.J.; Li, Y.T.; Zou, W.; Wang, L.; Pei, X.F.; Xie, M.; Huang, W.H. Simultaneous generation of gradients with gradually changed slope in a microfluidic device for quantifying axon response. Anal. Chem. 2013, 85, 7842–7850. [Google Scholar] [CrossRef] [PubMed]
- Joanne, W.C.; Li, X.; Lin, B.; Shim, S.; Ming, G.-L.; Levchenko, A. A microfluidics-based turning assay reveals complex growth cone responses to integrated gradients of substrate-bound ECM molecules and diffusible guidance cues. Lab Chip 2008, 8, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Jiang, L.; Li, S.; Deng, J.; Li, Y.; Yao, J.; Li, B.; Zheng, J. Using microfluidic chip to form brain-derived neurotrophic factor concentration gradient for studying neuron axon guidance. Biomicrofluidics 2014, 8, 014108. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Menon, S.; Gupton, S.L. Passive microfluidic chamber for long-term imaging of axon guidance in response to soluble gradients. Lab Chip 2015, 15, 2781–2789. [Google Scholar] [CrossRef] [PubMed]
- Kothapalli, C.R.; van Veen, E.; de Valence, S.; Chung, S.; Zervantonakis, I.K.; Gertler, F.B.; Kamm, R.D. A high-throughput microfluidic assay to study neurite response to growth factor gradients. Lab Chip 2011, 11, 497–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, A.M.; Blurton-Jones, M.; Rhee, S.W.; Cribbs, D.H.; Cotman, C.W.; Jeon, N.L. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat. Methods 2005, 2, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Dieterich, D.C.; Ito, H.T.; Kim, S.A.; Schuman, E.M. Microfluidic local perfusion chambers for the visualization and manipulation of synapses. Neuron 2010, 66, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Hellman, A.N.; Vahidi, B.; Kim, H.J.; Mismar, W.; Steward, O.; Jeon, N.L.; Venugopalan, V. Examination of axonal injury and regeneration in micropatterned neuronal culture using pulsed laser microbeam dissection. Lab Chip 2010, 10, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Vahidi, B.; Kim, H.J.; Rhee, S.W.; Jeon, N.L. Quantitative analysis of CNS axon regeneration using a microfluidic neuron culture device. Biochip J. 2008, 2, 44–51. [Google Scholar]
- Gao, Y.; Broussard, J.; Haque, A.; Revzin, A.; Lin, T. Functional imaging of neuron–astrocyte interactions in a compartmentalized microfluidic device. Microsyst. Nanoeng. 2016, 2, 15045. [Google Scholar] [CrossRef]
- Johnstone, A.F.M.; Gross, G.W.; Weiss, D.G.; Schroeder, O.H.U.; Gramowski, A.; Shafer, T.J. Microelectrode arrays: A physiologically based neurotoxicity testing platform for the 21st century. Neurotoxicology 2010, 31, 331–350. [Google Scholar] [CrossRef] [PubMed]
- Dworak, B.J.; Wheeler, B.C. Novel MEA platform with PDMS microtunnels enables the detection of action potential propagation from isolated axons in culture. Lab Chip 2009, 9, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Ravula, S.K.; Wang, M.S.; McClain, M.A.; Asress, S.A.; Frazier, B.; Glass, J.D. Spatiotemporal localization of injury potentials in DRG neurons during vincristine-induced axonal degeneration. Neurosci. Lett. 2007, 415, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Biffi, E.; Piraino, F.; Pedrocchi, A.; Fiore, G.B.; Ferrigno, G.; Redaelli, A.; Menegon, A.; Rasponi, M. A microfluidic platform for controlled biochemical stimulation of twin neuronal networks. Biomicrofluidics 2012, 6. [Google Scholar] [CrossRef] [PubMed]
- Biffi, E.; Menegon, A.; Piraino, F.; Pedrocchi, A.; Fiore, G.B.; Rasponi, M. Validation of long-term primary neuronal cultures and network activity through the integration of reversibly bonded microbioreactors and MEA substrates. Biotechnol. Bioeng. 2012, 109, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.; Weir, K.; Easton, C.; Huynh, W.; Moody, W.J.; Folch, A. A microfluidic microelectrode array for simultaneous electrophysiology, chemical stimulation, and imaging of brain slices. Lab Chip 2013, 13, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Drug transport across the blood–brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Lockman, P.R.; Mumper, R.J.; Khan, M.A.; Allen, D.D. Nanoparticle technology for drug delivery across the blood-brain barrier. Drug Dev. Ind. Pharm. 2002, 28, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Griep, L.M.; Wolbers, F.; de Wagenaar, B.; Ter Braak, P.M.; Weksler, B.B.; Romero, I.A.; Couraud, P.O.; Vermes, I.; van der Meer, A.D.; van den Berg, A. BBB on CHIP: Microfluidic platform to mechanically and biochemically modulate blood-brain barrier function. Biomed. Microdevices 2013, 15, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Prabhakarpandian, B.; Shen, M.-C.; Nichols, J.B.; Mills, I.R.; Sidoryk-Wegrzynowicz, M.; Aschner, M.; Pant, K. SyM-BBB: A microfluidic blood brain barrier model. Lab Chip 2013, 13, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Achyuta, A.K.H.; Conway, A.J.; Crouse, R.B.; Bannister, E.C.; Lee, R.N.; Katnik, C.P.; Behensky, A.A.; Cuevas, J.; Sundaram, S.S. A modular approach to create a neurovascular unit-on-a-chip. Lab Chip 2013, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Booth, R.; Kim, H. Characterization of a microfluidic in vitro model of the blood-brain barrier (μBBB). Lab Chip 2012, 12, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Pensabene, V.; Markov, D.A.; Allwardt, V.; Diana Neely, M.; Shi, M.; Britt, C.M.; Hoilett, O.S.; Yang, Q.; Brewer, B.M.; et al. Recreating blood-brain barrier physiology and structure on chip: A novel neurovascular microfluidic bioreactor. Biomicrofluidics 2015, 9, 054124. [Google Scholar] [CrossRef] [PubMed]
- Herland, A.; van der Meer, A.D.; FitzGerald, E.A.; Park, T.E.; Sleeboom, J.J.F.; Ingber, D.E. Distinct contributions of astrocytes and pericytes to neuroinflammation identified in a 3D human blood-brain barrier on a chip. PLoS ONE 2016, 11, e0150360. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.D.; Khafagy, E.S.; Khanafer, K.; Takayama, S.; Elsayed, M.E.H. Organization of endothelial cells, pericytes, and astrocytes into a 3D microfluidic in vitro model of the blood-brain barrier. Mol. Pharm. 2016, 13, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Seo, J.H.; Wong, K.H.K.; Terasaki, Y.; Park, J.; Bong, K.; Arai, K.; Lo, E.H.; Irimia, D. Three-dimensional blood-brain barrier model for in vitro studies of neurovascular pathology. Sci. Rep. 2015, 5, 15222. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.; Lan, F.; Lee, A.S.; Gong, T.; Sanchez-Freire, V.; Wang, Y.; Diecke, S.; Sallam, K.; Knowles, J.W.; Wang, P.J.; et al. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation 2013, 127, 1677–1691. [Google Scholar] [CrossRef] [PubMed]
- Ugolini, G.S.; Rasponi, M.; Pavesi, A.; Santoro, R.; Kamm, R.; Fiore, G.B.; Pesce, M.; Soncini, M. On-chip assessment of human primary cardiac fibroblasts proliferative responses to uniaxial cyclic mechanical strain. Biotechnol. Bioeng. 2016, 113, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Marsano, A.; Conficconi, C.; Lemme, M.; Occhetta, P.; Gaudiello, E.; Votta, E.; Cerino, G.; Redaelli, A.; Rasponi, M. Beating heart on a chip: A novel microfluidic platform to generate functional 3D cardiac microtissues. Lab Chip 2015, 16, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Pavesi, A.; Adriani, G.; Rasponi, M.; Zervantonakis, I.; Fiore, G.; Kamm, R. Controlled electromechanical cell stimulation on-a-chip. Sci. Rep. 2015, 5, 11800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abassi, Y.A.; Xi, B.; Li, N.; Ouyang, W.; Seiler, A.; Watzele, M.; Kettenhofen, R.; Bohlen, H.; Ehlich, A.; Kolossov, E.; et al. Dynamic monitoring of beating periodicity of stem cell-derived cardiomyocytes as a predictive tool for preclinical safety assessment. Br. J. Pharmacol. 2012, 165, 1424–1441. [Google Scholar] [CrossRef] [PubMed]
- Xi, B.; Wang, T.; Li, N.; Ouyang, W.; Zhang, W.; Wu, J.; Xu, X.; Wang, X.; Abassi, Y.A. Functional cardiotoxicity profiling and screening using the xCELLigence RTCA Cardio System. J. Lab. Autom. 2011, 16, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Wang, T.; Wang, Q.; Zhou, J.; Zou, L.; Su, K.; Wu, J.; Wang, P. High-performance beating pattern function of human induced pluripotent stem cell-derived cardiomyocyte-based biosensors for hERG inhibition recognition. Biosens. Bioelectron. 2015, 67, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Hu, N.; Cao, J.; Wu, J.; Su, K.; Wang, P. A cardiomyocyte-based biosensor for antiarrhythmic drug evaluation by simultaneously monitoring cell growth and beating. Biosens. Bioelectron. 2013, 49, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Werdich, A.A.; Lima, E.A.; Ivanov, B.; Ges, I.; Anderson, M.E.; Wikswo, J.P.; Baudenbacher, F.J. A microfluidic device to confine a single cardiac myocyte in a sub-nanoliter volume on planar microelectrodes for extracellular potential recordings. Lab Chip 2004, 4, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Ganitkevich, V.; Reil, S.; Schwethelm, B.; Schroeter, T.; Benndorf, K. Dynamic responses of single cardiomyocytes to graded ischemia studied by oxygen clamp in on-chip picochambers. Circ. Res. 2006, 99, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Klauke, N.; Smith, G.; Cooper, J.M. Microfluidic cell arrays for metabolic monitoring of stimulated cardiomyocytes. Electrophoresis 2010, 31, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Grosberg, A.; Alford, P.W.; McCain, M.L.; Parker, K.K. Ensembles of engineered cardiac tissues for physiological and pharmacological study: Heart on a chip. Lab Chip 2011, 11, 4165–4173. [Google Scholar] [CrossRef] [PubMed]
- Heidi Au, H.T.; Cui, B.; Chu, Z.E.; Veres, T.; Radisic, M. Cell culture chips for simultaneous application of topographical and electrical cues enhance phenotype of cardiomyocytes. Lab Chip 2009, 9, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Goss, J.A.; Cho, A.; McCain, M.L.; Parker, K.K. Microfluidic heart on a chip for higher throughput pharmacological studies. Lab Chip 2013, 13, 3599–3608. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; McCain, M.L.; Yang, L.; He, A.; Pasqualini, F.S.; Agarwal, A.; Yuan, H.; Jiang, D.; Zhang, D.; Zangi, L.; et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 2014, 20, 616–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, S.S.; Miklas, J.W.; Liu, J.; Aschar-Sobbi, R.; Xiao, Y.; Zhang, B.; Jiang, J.; Massé, S.; Gagliardi, M.; Hsieh, A.; et al. Biowire: A platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nat. Methods 2013, 10, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Nunes, S.S. Biowire platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Methods 2016, 101, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Zhang, B.; Liu, H.; Miklas, J.W.; Gagliardi, M.; Pahnke, A.; Thavandiran, N.; Sun, Y.; Simmons, C.; Keller, G.; et al. Microfabricated perfusable cardiac biowire: A platform that mimics native cardiac bundle. Lab Chip 2014, 14, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Mathur, A.; Loskill, P.; Shao, K.; Huebsch, N.; Hong, S.; Marcus, S.G.; Marks, N.; Mandegar, M.; Conklin, B.R.; Lee, L.P.; et al. Human iPSC-based cardiac microphysiological system for drug screening applications. Sci. Rep. 2015, 5, 8883. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Hotaling, N.A.; Ku, D.N.; Forest, C.R. Microfluidic thrombosis under multiple shear rates and antiplatelet therapy doses. PLoS ONE 2014, 9, e82493. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Ku, D.N.; Forest, C.R. Microfluidic system for simultaneous optical measurement of platelet aggregation at multiple shear rates in whole blood. Lab Chip 2012, 12, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Dimasi, A.; Rasponi, M.; Sheriff, J.; Chiu, W.C.; Bluestein, D.; Tran, P.L.; Slepian, M.J.; Redaelli, A. Microfluidic emulation of mechanical circulatory support device shear-mediated platelet activation. Biomed. Microdevices 2015, 17, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korin, N.; Kanapathipillai, M.; Matthews, B.D.; Crescente, M.; Brill, A.; Mammoto, T.; Ghosh, K.; Jurek, S.; Bencherif, S.A.; Bhatta, D.; et al. Shear-activated nanotherapeutics for drug targeting to obstructed blood vessels. Science 2012, 337, 738–742. [Google Scholar] [CrossRef] [PubMed]
- Chrobak, K.M.; Potter, D.R.; Tien, J. Formation of perfused, functional microvascular tubes in vitro. Microvasc. Res. 2006, 71, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, S.; Chung, M.; Kim, J.H.; Jeon, N.L. A bioengineered array of 3D microvessels for vascular permeability assay. Microvasc. Res. 2014, 91, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Sasaki, N.; Ato, M.; Hirakawa, S.; Sato, K.; Sato, K. Microcirculation-on-a-chip: A microfluidic platform for assaying blood- and lymphatic-vessel permeability. PLoS ONE 2015, 10, e0137301. [Google Scholar] [CrossRef] [PubMed]
- Günther, A.; Yasotharan, S.; Vagaon, A.; Lochovsky, C.; Pinto, S.; Yang, J.; Lau, C.; Voigtlaender-Bolz, J.; Bolz, S.-S. A microfluidic platform for probing small artery structure and function. Lab Chip 2010, 10, 2341–2349. [Google Scholar] [CrossRef] [PubMed]
- Yasotharan, S.; Pinto, S.; Sled, J.G.; Bolz, S.-S.; Günther, A. Artery-on-a-chip platform for automated, multimodal assessment of cerebral blood vessel structure and function. Lab Chip 2015, 15, 2660–2669. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W.R.; Griffith, B.P. Reconstructing the lung. Science 2010, 329, 520–522. [Google Scholar] [CrossRef] [PubMed]
- Hittinger, M.; Juntke, J.; Kletting, S.; Schneider-Daum, N.; de Souza Carvalho, C.; Lehr, C.-M. Preclinical safety and efficacy models for pulmonary drug delivery of antimicrobials with focus on in vitro models. Adv. Drug Deliv. Rev. 2015, 85, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.A.; Vanbever, R. Preclinical models for pulmonary drug delivery. Expert Opin. Drug Deliv. 2009, 6, 1231–1245. [Google Scholar] [CrossRef] [PubMed]
- Hakkinen, K.M.; Harunaga, J.S.; Doyle, A.D.; Yamada, K.M. Direct comparisons of the morphology, migration, cell adhesions, and actin cytoskeleton of fibroblasts in four different three-dimensional extracellular matrices. Tissue Eng. Part A 2011, 17, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Capulli, A.K.; Tian, K.; Mehandru, N.; Bukhta, A.; Choudhury, S.F.; Suchyta, M.; Parker, K.K. Approaching the in vitro clinical trial: Engineering organs on chips. Lab Chip 2014, 14, 3181–3186. [Google Scholar] [CrossRef] [PubMed]
- Zosky, G.R.; Sly, P.D. Animal models of asthma. Clin. Exp. Allergy 2007, 37, 973–988. [Google Scholar] [CrossRef] [PubMed]
- Van der Worp, H.B.; Howells, D.W.; Sena, E.S.; Porritt, M.J.; Rewell, S.; O’Collins, V.; Macleod, M.R. Can animal models of disease reliably inform human studies? PLoS Med. 2010, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nichols, J.E.; Niles, J.A.; Vega, S.P.; Argueta, L.B.; Eastaway, A.; Cortiella, J. Modeling the lung: Design and development of tissue engineered macro- and micro-physiologic lung models for research use. Exp. Biol. Med. 2014, 239, 1135–1169. [Google Scholar] [CrossRef] [PubMed]
- Dongeun, H.; Benjamin, D.M.; Akiko, M.; Martín, M.-Z.; Hong, Y.H.; Donald, E.I. Reconstituting organ level lung functions on a chip. Science 2010, 328, 1662–1668. [Google Scholar]
- Huh, D.; Leslie, D.C.; Matthews, B.D.; Fraser, J.P.; Jurek, S.; Hamilton, G.A.; Thorneloe, K.S.; McAlexander, M.A.; Ingber, D.E. A human disease model of drug toxicity-induced pulmonary edema in a lung-on-a-chip microdevice. Sci. Transl. Med. 2012, 4, 159ra147. [Google Scholar] [CrossRef] [PubMed]
- Huh, D. A human breathing lung-on-a-chip. Ann. Am. Thorac. Soc. 2015, 12, S42–S44. [Google Scholar] [CrossRef] [PubMed]
- Benam, K.H.; Villenave, R.; Lucchesi, C.; Varone, A.; Hubeau, C.; Lee, H.-H.; Alves, S.E.; Salmon, M.; Ferrante, T.C.; Weaver, J.C.; et al. Small airway-on-a-chip enables analysis of human lung inflammation and drug responses in vitro. Nat. Methods 2016, 13, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Kudlacz, E.; Conklyn, M.; Andresen, C.; Whitney-Pickett, C.; Changelian, P. The JAK-3 inhibitor CP-690550 is a potent anti-inflammatory agent in a murine model of pulmonary eosinophilia. Eur. J. Pharmacol. 2008, 582, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Nesmith, A.P.; Agarwal, A.; McCain, M.L.; Parker, K.K. Human airway musculature on a chip: An in vitro model of allergic asthmatic bronchoconstriction and bronchodilation. Lab Chip 2014, 14, 3925–3936. [Google Scholar] [CrossRef] [PubMed]
- Chetta, A.; Foresi, A.; del Donno, M.; Bertorelli, G.; Pesci, A.; Olivieri, D. Airways remodeling is a distinctive feature of asthma and is related to severity of disease. Chest 1997, 111, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, L.B.; Fryer, A.D.; Hirshman, C.A. M2 muscarinic receptors inhibit isoproterenol-induced relaxation of canine airway smooth muscle. J. Pharmacol. Exp. Ther. 1992, 262, 119–126. [Google Scholar] [PubMed]
- Stucki, A.O.; Stucki, J.D.; Hall, S.R.R.; Felder, M.; Mermoud, Y.; Schmid, R.A.; Geiser, T.; Guenat, O.T. A lung-on-a-chip array with an integrated bio-inspired respiration mechanism. Lab Chip 2015, 15, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yue, F.; Zhang, L.; Wang, J.; Wang, Y.; Li, J.; Lin, B.; Qi, W. Application of microfluidic gradient chip in the analysis of lung cancer chemotherapy resistance. J. Pharm. Biomed. Anal. 2009, 49, 806–810. [Google Scholar]
- Ying, L.; Zhu, Z.; Xu, Z.; He, T.; Li, E.; Guo, Z.; Liu, F.; Jiang, C.; Wang, Q. Cancer associated fibroblast-derived hepatocyte growth factor inhibits the paclitaxel-induced apoptosis of lung cancer A549 cells by up-regulating the PI3K/Akt and GRP78 signaling on a microfluidic platform. PLoS ONE 2015, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hirschhaeuser, F.; Menne, H.; Dittfeld, C.; West, J.; Mueller-Klieser, W.; Kunz-Schughart, L.A. Multicellular tumor spheroids: An underestimated tool is catching up again. J. Biotechnol. 2010, 148, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Barbone, D.; Yang, T.M.; Morgan, J.R.; Gaudino, G.; Broaddus, V.C. Mammalian target of rapamycin contributes to the acquired apoptotic resistance of human mesothelioma multicellular spheroids. J. Biol. Chem. 2008, 283, 13021–13030. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, W.; Wang, Y.; Wang, J.; Tu, Q.; Liu, R.; Wang, J. Construction of oxygen and chemical concentration gradients in a single microfluidic device for studying tumor cell-drug interactions in a dynamic hypoxia microenvironment. Lab Chip 2013, 13, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Ruppen, J.; Cortes-Dericks, L.; Marconi, E.; Karoubi, G.; Schmid, R.A.; Peng, R.; Marti, T.M.; Guenat, O.T. A microfluidic platform for chemoresistive testing of multicellular pleural cancer spheroids. Lab Chip 2014, 14, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Gao, Y.; Hao, Y.; Li, E.; Wang, Y.; Zhang, J.; Wang, W.; Gao, Z.; Wang, Q. Application of a microfluidic chip-based 3D co-culture to test drug sensitivity for individualized treatment of lung cancer. Biomaterials 2013, 34, 4109–4117. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.-J.; Suh, K.-Y. A multi-layer microfluidic device for efficient culture and analysis of renal tubular cells. Lab Chip 2010, 10, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Pallone, T.L.; Silldorff, E.P.; Turner, M.R. Intrarenal blood flow: Microvascular anatomy and the regulation of medullary perfusion. Clin. Exp. Pharmacol. Physiol. 1998, 25, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Tiong, H.Y.; Huang, P.; Xiong, S.; Li, Y.; Vathsala, A.; Zink, D. Drug-induced nephrotoxicity: Clinical impact and preclinical in vitro models. Mol. Pharm. 2014, 11, 1933–1948. [Google Scholar] [CrossRef] [PubMed]
- Naughton, C.A. Drug-induced nephrotoxicity. Am. Fam. Phys. 2008, 78, 743–750. [Google Scholar]
- Perazella, M.A. Renal vulnerability to drug toxicity. Clin. J. Am. Soc. Nephrol. 2009, 4, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Schetz, M.; Dasta, J.; Goldstein, S.; Golper, T. Drug-induced acute kidney injury. Curr. Opin. Crit. Care 2005, 11, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Jenkinson, S.E.; Chung, G.W.; van Loon, E.; Bakar, N.S.; Dalzell, A.M.; Brown, C.D.A. The limitations of renal epithelial cell line HK-2 as a model of drug transporter expression and function in the proximal tubule. Pflugers Arch. Eur. J. Physiol. 2012, 464, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Snouber, L.C.; Letourneur, F.; Chafey, P.; Broussard, C.; Monge, M.; Legallais, C.; Leclerc, E. Analysis of transcriptomic and proteomic profiles demonstrates improved Madin-Darby canine kidney cell function in a renal microfluidic biochip. Biotechnol. Prog. 2012, 28, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Essig, M.; Terzi, F.; Burtin, M.; Friedlander, G. Mechanical strains induced by tubular flow affect the phenotype of proximal tubular cells. Am. J. Physiol. Ren. Physiol. 2001, 281, F751–F762. [Google Scholar]
- Bhat, V.D.; Windridge, P.A.; Cherry, R.S.; Mandel, L.J. Fluctuating shear stress effects on stress fiber architecture and energy metabolism of cultured renal cells. Biotechnol. Prog. 1995, 11, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Yan, Q.; Duan, Y.; Weinbaum, S.; Weinstein, A.M.; Wang, T. Axial flow modulates proximal tubule NHE3 and H-ATPase activities by changing microvillus bending moments. Am. J. Physiol. Ren. Physiol. 2006, 290, F289–F296. [Google Scholar] [CrossRef] [PubMed]
- Wilmer, M.J.; Ng, C.P.; Lanz, H.L.; Vulto, P.; Suter-Dick, L.; Masereeuw, R. Kidney-on-a-chip technology for drug-induced nephrotoxicity screening. Trends Biotechnol. 2016, 34, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, E.; Sakai, Y.; Fujii, T. Microfluidic PDMS (Polydimethylsiloxane) bioreactor for large-scale culture of hepatocytes. Biotechnol. Prog. 2004, 20, 750–755. [Google Scholar] [CrossRef] [PubMed]
- Baudoin, R.; Griscom, L.; Monge, M.; Legallais, C.; Leclerc, E. Development of a renal microchip for in vitro distal tubule models. Biotechnol. Prog. 2007, 23, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Marison, I.W.; von Stockar, U. The importance of ammonia in mammalian cell culture. J. Biotechnol. 1996, 46, 161–185. [Google Scholar] [CrossRef]
- Choucha Snouber, L.; Jacques, S.; Monge, M.; Legallais, C.; Leclerc, E. Transcriptomic analysis of the effect of ifosfamide on MDCK cells cultivated in microfluidic biochips. Genomics 2012, 100, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. The IkappaB kinase—A bridge between inflammation and cancer. Cell Res. 2008, 18, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Elwi, A.N.; Damaraju, V.L.; Kuzma, M.L.; Mowles, D.A.; Baldwin, S.A.; Young, J.D.; Sawyer, M.B.; Cass, C.E. ransepithelial fluxes of adenosine and 2′-deoxyadenosine across human renal proximal tubule cells: Roles of nucleoside transporters hENT1, hENT2, and hCNT. Am. J. Physiol. Ren. Physiol. 2009, 296, F1439–F1451. [Google Scholar] [CrossRef] [PubMed]
- Rebelo, L.; Carmo-Fonseca, M.; Moura, T.F. Redistribution of microvilli and membrane enzymes in isolated rat proximal tubule cells. Biol. Cell 1992, 74, 203–209. [Google Scholar] [CrossRef]
- Jang, K.-J.; Cho, H.S.; Kang, D.H.; Bae, W.G.; Kwon, T.-H.; Suh, K.-Y. Fluid-shear-stress-induced translocation of aquaporin-2 and reorganization of actin cytoskeleton in renal tubular epithelial cells. Integr. Biol. 2011, 3, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Ramello, C.; Paullier, P.; Ould-Dris, A.; Monge, M.; Legallais, C.; Leclerc, E. Investigation into modification of mass transfer kinetics by acrolein in a renal biochip. Toxicol. In Vitro 2011, 25, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; LesherPerez, S.C.; Kim, B.C.C.; Yamanishi, C.; Labuz, J.M.; Leung, B.; Takayama, S. Pharmacokinetic profile that reduces nephrotoxicity of gentamicin in a perfused kidney-on-a-chip. Biofabrication 2016, 8, 015021. [Google Scholar] [CrossRef] [PubMed]
- Mavros, M.N.; Polyzos, K.A.; Rafailidis, P.I.; Falagas, M.E. Once versus multiple daily dosing of aminoglycosides for patients with febrile neutropenia: A systematic review and meta-analysis. J. Antimicrob. Chemother. 2011, 66, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Gotoh, N.; Yan, Q.; Du, Z.; Weinstein, A.M.; Wang, T.; Weinbaum, S. Shear-induced reorganization of renal proximal tubule cell actin cytoskeleton and apical junctional complexes. Proc. Natl. Acad. Sci. USA 2008, 105, 11418–11423. [Google Scholar] [CrossRef] [PubMed]
- Andes, D.; Craig, W.A. Animal model pharmacokinetics and pharmacodynamics: A critical review. Int. J. Antimicrob. Agents 2002, 19, 261–268. [Google Scholar] [CrossRef]
- Paine, S.W.; Ménochet, K.; Denton, R.; McGinnity, D.F.; Riley, R.J. Prediction of human renal clearance from preclinical species for a diverse set of drugs that exhibit both active secretion and net reabsorption. Drug Metab. Dispos. 2011, 39, 1008–1013. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Bleasby, K.; Evers, R. Species differences in drug transporters and implications for translating preclinical findings to humans. Expert Opin. Drug Metab. Toxicol. 2012, 5255, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Pfaller, W.; Gstraunthaler, G. Nephrotoxicity testing in vitro—What we know and what we need to know. Environ. Health Perspect. 1998, 106 (Suppl. 2), 559–569. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.-J.; Mehr, A.P.; Hamilton, G.A.; McPartlin, L.A.; Chung, S.; Suh, K.-Y.; Ingber, D.E. Human kidney proximal tubule-on-a-chip for drug transport and nephrotoxicity assessment. Integr. Biol. (Camb.) 2013, 5, 1119–1129. [Google Scholar] [CrossRef] [PubMed]
- Adler, M.; Ramm, S.; Hafner, M.; Muhlich, J.L.; Gottwald, E.M.; Weber, E.; Jaklic, A.; Ajay, A.K.; Svoboda, D.; Auerbach, S.; et al. A quantitative approach to screen for nephrotoxic compounds in vitro. J. Am. Soc. Nephrol. 2016, 27, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.J.; Wang, Z.; Voellinger, J.L.; Yeung, C.K.; Shen, D.D.; Thummel, K.E.; Zheng, Y.; Ligresti, G.; Eaton, D.L.; Muczynski, K.A.; et al. Innovations in preclinical biology: Ex vivo engineering of a human kidney tissue microperfusion system. Stem Cell Res. Ther. 2013, 4 (Suppl. 1), S17. [Google Scholar] [CrossRef] [PubMed]
- Weber, E.J.; Chapron, A.; Chapron, B.D.; Voellinger, J.L.; Lidberg, K.A.; Yeung, C.K.; Wang, Z.; Yamaura, Y.; Hailey, D.W.; Neumann, T.; et al. Development of a microphysiological model of human kidney proximal tubule function. Kidney Int. 2016, 90, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Madden, L.; Juhas, M.; Kraus, W.E.; Truskey, G.A.; Bursac, N. Bioengineered human myobundles mimic clinical responses of skeletal muscle to drugs. eLife 2015, 4, e04885. [Google Scholar] [CrossRef] [PubMed]
- Occhetta, P.; Centola, M.; Tonnarelli, B.; Redaelli, A.; Martin, I.; Rasponi, M. High-throughput microfluidic platform for 3D cultures of mesenchymal stem cells, towards engineering developmental processes. Sci. Rep. 2015, 5, 10288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodson, K.H.; Echevarria, F.D.; Li, D.; Sappington, R.M.; Edd, J.F. Retina-on-a-chip: A microfluidic platform for point access signaling studies. Biomed. Microdevices 2015, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rigat-Brugarolas, L.G.; Elizalde-Torrent, A.; Bernabeu, M.; de Niz, M.; Martin-Jaular, L.; Fernandez-Becerra, C.; Homs-Corbera, A.; Samitier, J.; del Portillo, H.A. A functional microengineered model of the human splenon-on-a-chip. Lab Chip 2014, 14, 1715–1724. [Google Scholar] [CrossRef] [PubMed]
- Clavica, F.; Homsy, A.; Jeandupeux, L.; Obrist, D. Red blood cell phase separation in symmetric and asymmetric microchannel networks: Effect of capillary dilation and inflow velocity. Sci. Rep. 2016, 6, 36763. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.Q.; Park, W.T. Rapid, low-cost fabrication of circular microchannels by air expansion into partially cured polymer. Sens. Actuators B Chem. 2016, 235, 302–308. [Google Scholar] [CrossRef]
- Maschmeyer, I.; Lorenz, A.K.; Schimek, K.; Hasenberg, T.; Ramme, A.P.; Hübner, J.; Lindner, M.; Drewell, C.; Bauer, S.; Thomas, A.; et al. A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents. Lab Chip 2015, 15, 2688–2699. [Google Scholar] [CrossRef] [PubMed]
- Materne, E.M.; Ramme, A.P.; Terrasso, A.P.; Serra, M.; Alves, P.M.; Brito, C.; Sakharov, D.A.; Tonevitsky, A.G.; Lauster, R.; Marx, U. A multi-organ chip co-culture of neurospheres and liver equivalents for long-term substance testing. J. Biotechnol. 2014, 205, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Vunjak-Novakovic, G.; Bhatia, S.; Chen, C.; Hirschi, K. HeLiVa platform: Integrated heart-liver-vascular systems for drug testing in human health and disease. Stem Cell Res. Ther. 2013, 4, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Midwoud, P.M.; Merema, M.T.; Verpoorte, E.; Groothuis, G.M.M. A microfluidic approach for in vitro assessment of interorgan interactions in drug metabolism using intestinal and liver slices. Lab Chip 2010, 10, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Wagner, I.; Materne, E.-M.; Brincker, S.; Süssbier, U.; Frädrich, C.; Busek, M.; Sonntag, F.; Sakharov, D.A.; Trushkin, E.V.; Tonevitsky, A.G.; et al. A dynamic multi-organ-chip for long-term cultivation and substance testing proven by 3D human liver and skin tissue co-culture. Lab Chip 2013, 13, 3538–3547. [Google Scholar] [CrossRef] [PubMed]
- Marx, U.; Walles, H.; Hoffmann, S.; Lindner, G.; Horland, R.; Sonntag, F.; Klotzbach, U.; Sakharov, D.; Tonevitsky, A.; Lauster, R. “Human-on-a-chip” developments: A translational cuttingedge alternative to systemic safety assessment and efficiency evaluation of substances in laboratory animals and man? ATLA Altern. Lab. Anim. 2012, 40, 235–257. [Google Scholar] [PubMed]
- Sung, J.H.; Esch, M.B.; Prot, J.-M.; Long, C.J.; Smith, A.; Hickman, J.J.; Shuler, M.L. Microfabricated mammalian organ systems and their integration into models of whole animals and humans. Lab Chip 2013, 13, 1201–1212. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Kam, C.; Shuler, M.L. A microfluidic device for a pharmacokinetic-pharmacodynamic (PK-PD) model on a chip. Lab Chip 2010, 10, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Wikswo, J.P.; Curtis, E.L.; Eagleton, Z.E.; Evans, B.C.; Kole, A.; Hofmeister, L.H.; Matloff, W.J. Scaling and systems biology for integrating multiple organs-on-a-chip. Lab Chip 2013, 13, 3496–3511. [Google Scholar] [CrossRef] [PubMed]
- Moraes, C.; Labuz, J.M.; Leung, B.M.; Inoue, M.; Chun, T.-H.; Takayama, S. On being the right size: Scaling effects in designing a human-on-a-chip. Integr. Biol. 2013, 5, 1149–1161. [Google Scholar] [CrossRef] [PubMed]
- Viravaidya, K.; Sin, A.; Shuler, M.L. Development of a microscale cell culture analog to probe naphthalene toxicity. Biotechnol. Prog. 2004, 20, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Abaci, H.E.; Shuler, M.L. Human-on-a-chip design strategies and principles for physiologically based pharmacokinetics/pharmacodynamics modeling. Integr. Biol. 2015, 7, 383–391. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ugolini, G.S.; Cruz-Moreira, D.; Visone, R.; Redaelli, A.; Rasponi, M. Microfabricated Physiological Models for In Vitro Drug Screening Applications. Micromachines 2016, 7, 233. https://doi.org/10.3390/mi7120233
Ugolini GS, Cruz-Moreira D, Visone R, Redaelli A, Rasponi M. Microfabricated Physiological Models for In Vitro Drug Screening Applications. Micromachines. 2016; 7(12):233. https://doi.org/10.3390/mi7120233
Chicago/Turabian StyleUgolini, Giovanni Stefano, Daniela Cruz-Moreira, Roberta Visone, Alberto Redaelli, and Marco Rasponi. 2016. "Microfabricated Physiological Models for In Vitro Drug Screening Applications" Micromachines 7, no. 12: 233. https://doi.org/10.3390/mi7120233
APA StyleUgolini, G. S., Cruz-Moreira, D., Visone, R., Redaelli, A., & Rasponi, M. (2016). Microfabricated Physiological Models for In Vitro Drug Screening Applications. Micromachines, 7(12), 233. https://doi.org/10.3390/mi7120233