
Farewell to Animal Testing: Innovations on Human Intestinal Microphysiological Systems



Abstract
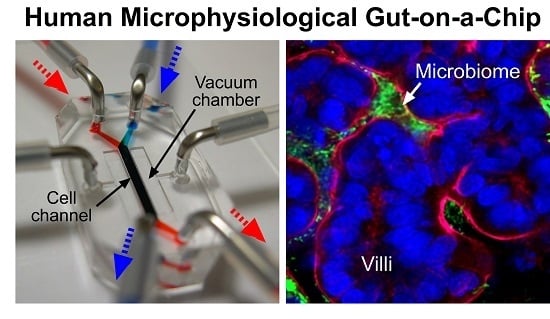
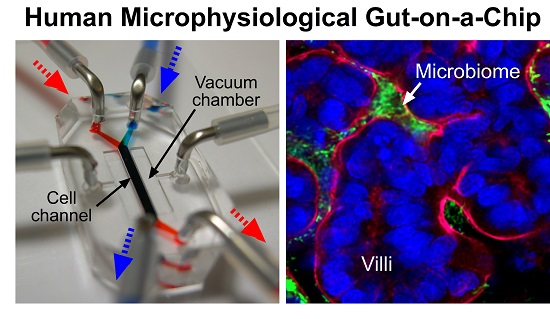
:
1. Introduction
1.1. Human Intestine, a Complex Organ to Mimic
1.2. Limitations in Animal Models
2. In Vitro Models to Mimic Tissue-Level Intestinal Pathophysiology
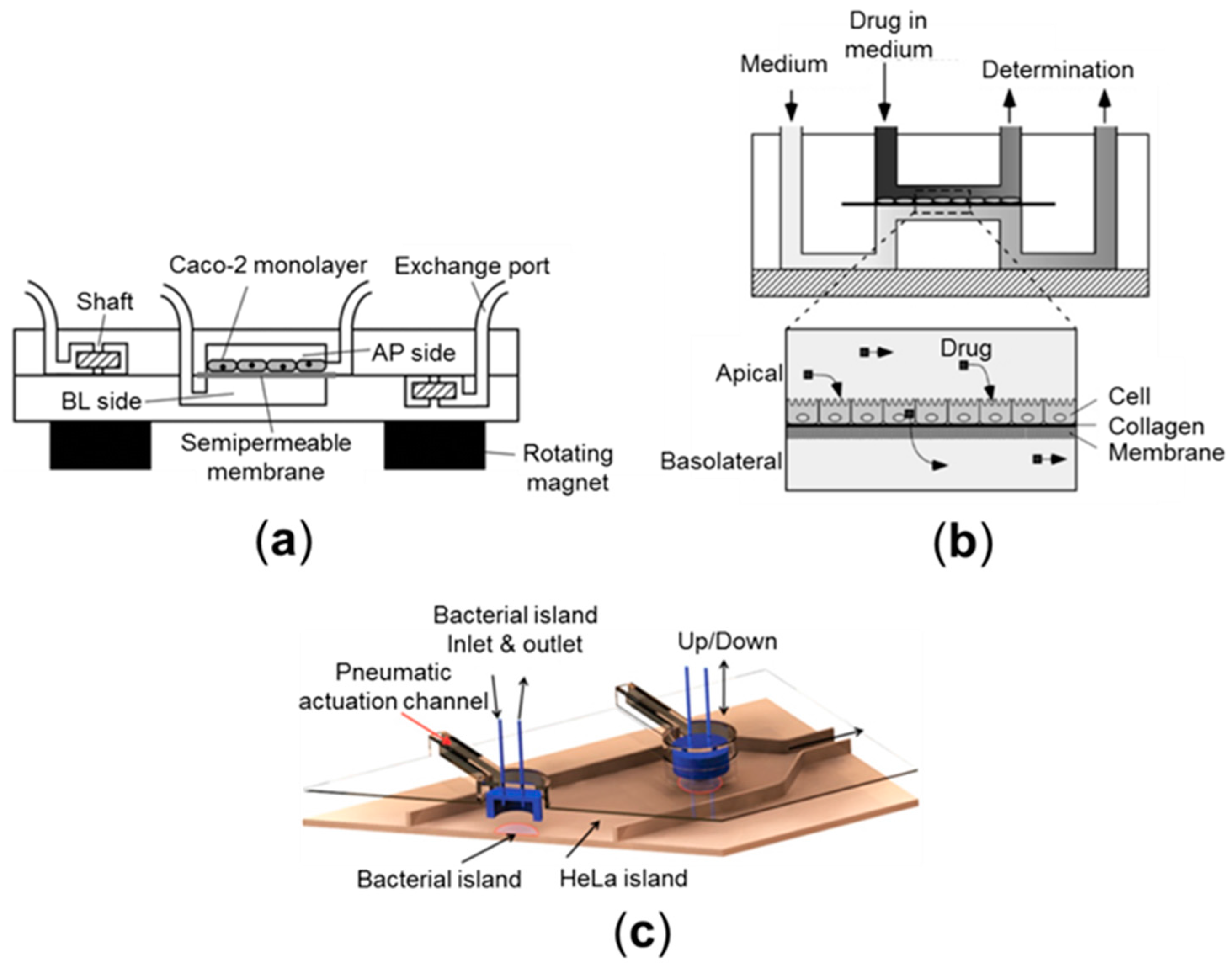
2.1. Cell Culture Models (2D)
2.2. Cell Culture Models (Pseudo-3D)
2.3. Organoid Culture Models (3D)
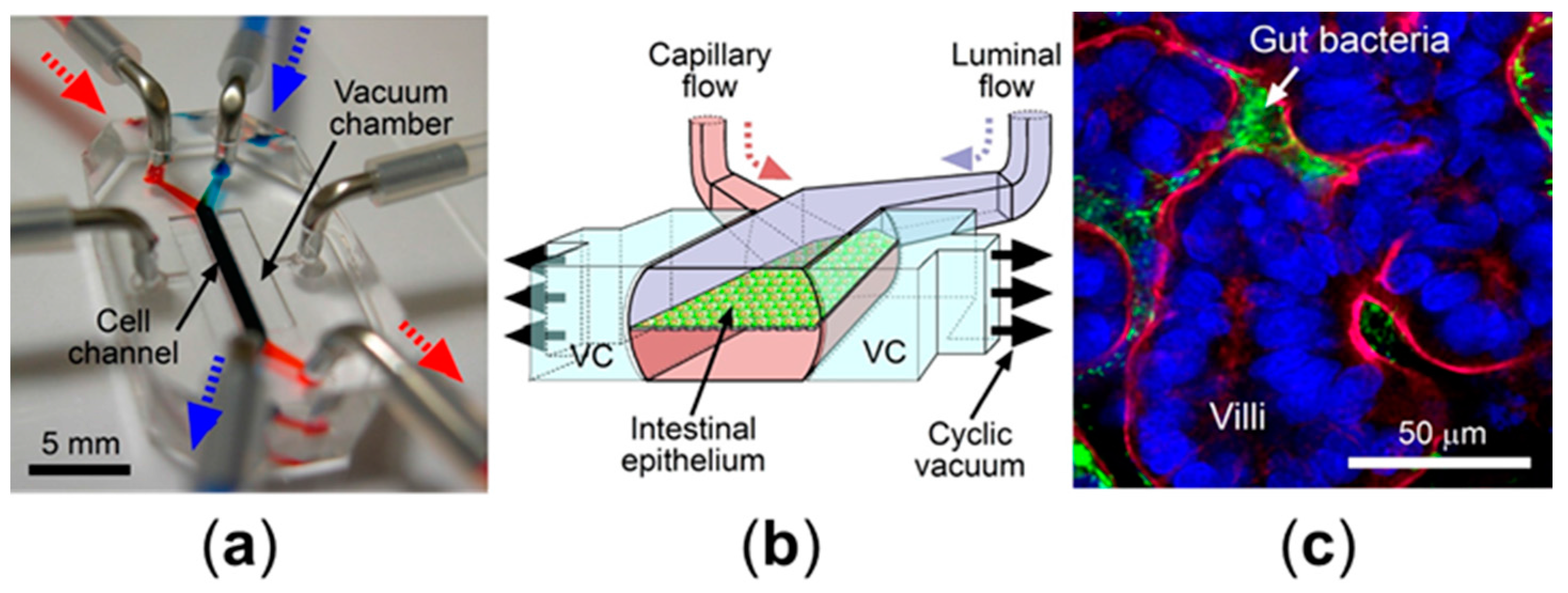
2.4. Microfluidic Culture Models
3. Human Gut-on-a-Chip: Emulating Organ-Level Intestinal Pathophysiology
3.1. Peristalsis
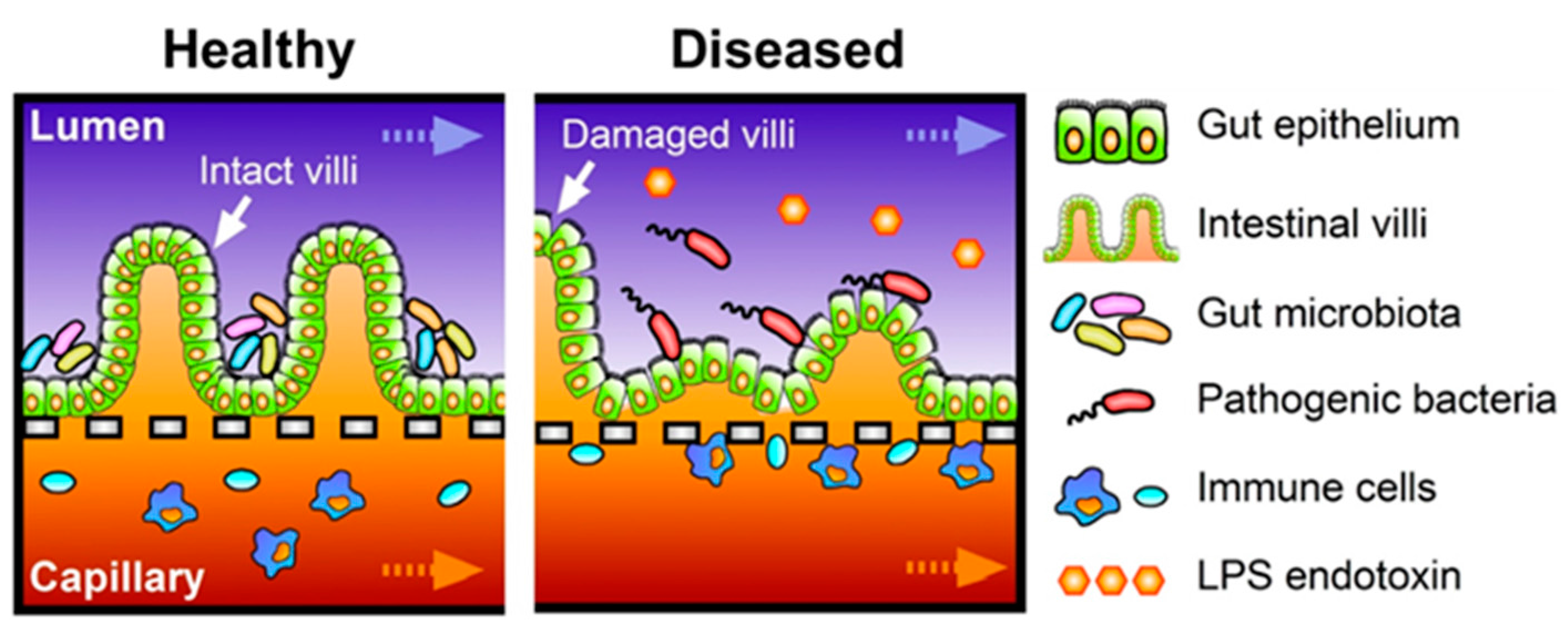
3.2. Host-Microbe Ecosystem
3.3. Immune Components
4. Prospects
4.1. Pharmaceutical Applications
4.2. Applications in the Food Industry
4.3. Revalidation of Probiotics and Prebiotics
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 2D | Two-dimensional |
| 3D | Three-dimensional |
| CD | Crohn’s disease |
| CRC | Colorectal cancer |
| DAPI | 4',6-diamidino-2-phenylindole |
| ECM | Extracellular matrix |
| EHEC | Enterohemorrhagic E. coli |
| EspP | EHEC serine protease |
| FcRn | Neonatal Fc receptor |
| GFP | Green-fluorescence-labeled protein |
| GALT | Gut-associated lymphoid tissues |
| IBD | Inflammatory bowel disease |
| IgG | Immunoglobulin G |
| IL | Interleukin |
| iPSCs | Induced pluripotent stem cells |
| LGG | Lactobacillus rhamnosus GG |
| LGR5 | Leucine-rich repeat-containing G-protein coupled receptor 5 |
| LPS | Lipopolysaccharide |
| MUC2 | Mucin 2 |
| NAS | Non-caloric artificial sweeteners |
| NHE3 | Na+/H+ exchanger 3 |
| PBMCs | Peripheral blood mononuclear cells |
| PD | Pharmacodynamics |
| PDMS | Polydimethylsiloxane |
| PK | Pharmacokinetics |
| SCFA | Short-chain fatty acids |
| SIBO | Small intestinal bacterial overgrowth |
| TEER | Trans-epithelial electrical resistance |
| TNF | Tumor necrosis factor |
| UC | Ulcerative colitis |
References
- Sommer, F.; Backhed, F. The gut microbiota—Masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Groschwitz, K.R.; Hogan, S.P. Intestinal barrier function: Molecular regulation and disease pathogenesis. J. Allergy Clin. Immun. 2009, 124, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Longman, R.S.; Littman, D.R. The functional impact of the intestinal microbiome on mucosal immunity and systemic autoimmunity. Curr. Opin. Rheumatol. 2015, 27, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Basson, M.D. Paradigms for mechanical signal transduction in the intestinal epithelium. Category: Molecular, cell, and developmental biology. Digestion 2003, 68, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Vantrappen, G.; Janssens, J.; Hellemans, J.; Ghoos, Y. The interdigestive motor complex of normal subjects and patients with bacterial overgrowth of the small intestine. J. Clin. Investig. 1977, 59, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Li, H.; Collins, J.J.; Ingber, D.E. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc. Natl. Acad. Sci. USA 2016, 113, E7–E15. [Google Scholar] [CrossRef] [PubMed]
- Maynard, C.L.; Elson, C.O.; Hatton, R.D.; Weaver, C.T. Reciprocal interactions of the intestinal microbiota and immune system. Nature 2012, 489, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Ohama, T.; Hori, M.; Ozaki, H. Mechanism of abnormal intestinal motility in inflammatory bowel disease: How smooth muscle contraction is reduced? J. Smooth Muscle Res. 2007, 43, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Gayer, C.P.; Basson, M.D. The effects of mechanical forces on intestinal physiology and pathology. Cell. Signal. 2009, 21, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Madl, C.; Druml, W. Gastrointestinal disorders of the critically ill. Systemic consequences of ileus. Best Pract. Res. Clin. Gastroenterol. 2003, 17, 445–456. [Google Scholar] [CrossRef]
- Mukhopadhya, I.; Hansen, R.; El-Omar, E.M.; Hold, G.L. IBD-what role do Proteobacteria play? Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.A.; Vanbever, R. Preclinical models for pulmonary drug delivery. Expert Opin. Drug Deliv. 2009, 6, 1231–1245. [Google Scholar] [CrossRef] [PubMed]
- Patterson, D.M.; Shohet, J.M.; Kim, E.S. Preclinical models of pediatric solid tumors (neuroblastoma) and their use in drug discovery. Curr. Protoc. Pharmacol. 2011. [Google Scholar] [CrossRef]
- Amit, D.; Gofrit, O.N.; Matouk, I.; Birman, T.; Hochberg, A. Use of preclinical models to assess the therapeutic potential of new drug candidates for bladder cancer. Semin. Oncol. 2012, 39, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R. The role of preclinical animal models in breast cancer drug development. Breast Cancer Res. 2009, 11, S22. [Google Scholar] [CrossRef] [PubMed]
- Rowland, M. Influence of route of administration on drug availability. J. Pharm. Sci. 1972, 61, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.M.; Tozer, T.N. First-pass elimination. Basic concepts and clinical consequences. Clin. Pharmacokinet. 1984, 9, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Z.; Nikaido, H. Efflux-mediated drug resistance in bacteria: An update. Drugs 2009, 69, 1555–1623. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.J.; Kim, H.G.; Kim, J.S.; Oh, D.G.; Um, Y.J.; Seo, C.S.; Han, J.W.; Cho, H.J.; Kim, G.H.; Jeong, T.C.; et al. The effect of gut microbiota on drug metabolism. Expert Opin. Drug Metab. Toxicol. 2013, 9, 1295–1308. [Google Scholar] [PubMed]
- Hintze, K.J.; Cox, J.E.; Rompato, G.; Benninghoff, A.D.; Ward, R.E.; Broadbent, J.; Lefevre, M. Broad scope method for creating humanized animal models for animal health and disease research through antibiotic treatment and human fecal transfer. Gut Microbes 2014, 5, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef] [PubMed]
- Engelman, R.W.; Kerr, W.G. Assessing inflammatory disease at mucosal surfaces in murine genetic models. Methods Mol. Biol. 2012, 900, 433–441. [Google Scholar] [PubMed]
- Rongvaux, A.; Willinger, T.; Martinek, J.; Strowig, T.; Gearty, S.V.; Teichmann, L.L.; Saito, Y.; Marches, F.; Halene, S.; Palucka, A.K.; et al. Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol. 2014, 32, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, D.C.; Carding, S.R. Inflammatory bowel disease: Cause and immunobiology. Lancet 2007, 369, 1627–1640. [Google Scholar] [CrossRef]
- Balimane, P.V.; Chong, S. Cell culture-based models for intestinal permeability: A critique. Drug Discov. Today 2005, 10, 335–343. [Google Scholar] [CrossRef]
- Hubatsch, I.; Ragnarsson, E.G.; Artursson, P. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat. Protoc. 2007, 2, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Haller, D.; Bode, C.; Hammes, W.P.; Pfeifer, A.M.; Schiffrin, E.J.; Blum, S. Non-pathogenic bacteria elicit a differential cytokine response by intestinal epithelial cell/leucocyte co-cultures. Gut 2000, 47, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Mahler, G.J.; Esch, M.B.; Tako, E.; Southard, T.L.; Archer, S.D.; Glahn, R.P.; Shuler, M.L. Oral exposure to polystyrene nanoparticles affects iron absorption. Nat. Nanotechnol. 2012, 7, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Huh, D.; Hamilton, G.; Ingber, D.E. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip 2012, 12, 2165–2174. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Ingber, D.E. Gut-on-a-chip microenvironment induces human intestinal cells to undergo villus differentiation. Integr. Biol. 2013, 5, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H.K. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 2007, 8, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Murthy, S.K.; Barabino, G.A.; Carrier, R.L. Synergic effects of crypt-like topography and ECM proteins on intestinal cell behavior in collagen based membranes. Biomaterials 2010, 31, 7586–7598. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Peng, S.; Luo, D.; March, J.C. In vitro 3D human small intestinal villous model for drug permeability determination. Biotechnol. Bioeng. 2012, 109, 2173–2178. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Murthy, S.K.; Fowle, W.H.; Barabino, G.A.; Carrier, R.L. Influence of micro-well biomimetic topography on intestinal epithelial Caco-2 cell phenotype. Biomaterials 2009, 30, 6825–6834. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Yu, J.; Luo, D.; Shuler, M.L.; March, J.C. Microscale 3-D hydrogel scaffold for biomimetic gastrointestinal (GI) tract model. Lab Chip 2011, 11, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Costello, C.M.; Hongpeng, J.; Shaffiey, S.; Yu, J.; Jain, N.K.; Hackam, D.; March, J.C. Synthetic small intestinal scaffolds for improved studies of intestinal differentiation. Biotechnol. Bioeng. 2014, 111, 1222–1232. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Clevers, H. Leucine-rich repeat-containing g-protein-coupled receptors as markers of adult stem cells. Gastroenterology 2010, 138, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Clevers, H. Growing self-organizing mini-guts from a single intestinal stem cell: Mechanism and applications. Science 2013, 340, 1190–1194. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Spence, J.R.; Mayhew, C.N.; Rankin, S.A.; Kuhar, M.F.; Vallance, J.E.; Tolle, K.; Hoskins, E.E.; Kalinichenko, V.V.; Wells, S.I.; Zorn, A.M.; et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 2011, 470, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Brugmann, S.A.; Wells, J.M. Building additional complexity to in vitro-derived intestinal tissues. Stem Cell Res. Ther. 2013, 4, S1. [Google Scholar] [CrossRef] [PubMed]
- Leslie, J.L.; Huang, S.; Opp, J.S.; Nagy, M.S.; Kobayashi, M.; Young, V.B.; Spence, J.R. Persistence and toxin production by clostridium difficile within human intestinal organoids result in disruption of epithelial paracellular barrier function. Infect. Immun. 2015, 83, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Engevik, M.A.; Engevik, K.A.; Yacyshyn, M.B.; Wang, J.; Hassett, D.J.; Darien, B.; Yacyshyn, B.R.; Worrell, R.T. Human clostridium difficile infection: Inhibition of NHE3 and microbiota profile. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G497–G509. [Google Scholar] [CrossRef] [PubMed]
- Engevik, M.A.; Yacyshyn, M.B.; Engevik, K.A.; Wang, J.; Darien, B.; Hassett, D.J.; Yacyshyn, B.R.; Worrell, R.T. Human clostridium difficile infection: Altered mucus production and composition. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G510–G524. [Google Scholar] [CrossRef] [PubMed]
- Stelzner, M.; Helmrath, M.; Dunn, J.C.; Henning, S.J.; Houchen, C.W.; Kuo, C.; Lynch, J.; Li, L.; Magness, S.T.; Martin, M.G.; et al. A nomenclature for intestinal in vitro cultures. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1359–G1363. [Google Scholar] [CrossRef] [PubMed]
- Finkbeiner, S.R.; Zeng, X.L.; Utama, B.; Atmar, R.L.; Shroyer, N.F.; Estes, M.K. Stem cell-derived human intestinal organoids as an infection model for rotaviruses. MBio 2012, 3, e00159–e00112. [Google Scholar] [CrossRef] [PubMed]
- Straub, T.M.; Honer zu Bentrup, K.; Orosz-Coghlan, P.; Dohnalkova, A.; Mayer, B.K.; Bartholomew, R.A.; Valdez, C.O.; Bruckner-Lea, C.J.; Gerba, C.P.; Abbaszadegan, M.; et al. In vitro cell culture infectivity assay for human noroviruses. Emerg. Infect. Dis. 2007, 13, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; Wiegerinck, C.L.; de Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; de Winter-de Groot, K.M.; Brandsma, A.M.; de Jong, N.W.M.; Bijvelds, M.J.C.; Scholte, B.J.; et al. A functional cftr assay using primary cystic fibrosis intestinal organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Foulke-Abel, J.; In, J.; Kovbasnjuk, O.; Zachos, N.C.; Ettayebi, K.; Blutt, S.E.; Hyser, J.M.; Zeng, X.L.; Crawford, S.E.; Broughman, J.R.; et al. Human enteroids as an ex vivo model of host-pathogen interactions in the gastrointestinal tract. Exp. Biol. Med. 2014, 239, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- In, J.; Foulke-Abel, J.; Cole, R.N.; Kovbasnjuk, O. Sa1743 EHEC virulence factor espp cleaves actin binding protein fodrin to induce macropinocytosis of shiga toxins. Gastroenterology 2014, 146, S286. [Google Scholar] [CrossRef]
- Mehling, M.; Tay, S. Microfluidic cell culture. Curr. Opin. Biotechnol. 2014, 25, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Qin, D.; Xia, Y.; Whitesides, G.M. Soft lithography for micro- and nanoscale patterning. Nat. Protoc. 2010, 5, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Yamamoto, T.; Sakai, H.; Sakai, Y.; Fujii, T. An integrated microfluidic system for long-term perfusion culture and on-line monitoring of intestinal tissue models. Lab Chip 2008, 8, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Imura, Y.; Asano, Y.; Sato, K.; Yoshimura, E. A microfluidic system to evaluate intestinal absorption. Anal. Sci. 2009, 25, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Mahler, G.J.; Shuler, M.L.; Glahn, R.P. Characterization of Caco-2 and HT29-MTX cocultures in an in vitro digestion/cell culture model used to predict iron bioavailability. J. Nutr. Biochem. 2009, 20, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Esch, M.B.; Sung, J.H.; Yang, J.; Yu, C.; Yu, J.; March, J.C.; Shuler, M.L. On chip porous polymer membranes for integration of gastrointestinal tract epithelium with microfluidic ‘body-on-a-chip’ devices. Biomed. Microdevices 2012, 14, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Sarna, S.K.; Otterson, M.F. Small intestinal physiology and pathophysiology. Gastroenterol. Clin. N. Am. 1989, 18, 375–404. [Google Scholar] [PubMed]
- Kim, J.; Hegde, M.; Jayaraman, A. Co-culture of epithelial cells and bacteria for investigating host-pathogen interactions. Lab Chip 2010, 10, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Nguyen, N.T. A polymeric cell stretching device for real-time imaging with optical microscopy. Biomed. Microdevices 2013, 15, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Nguyen, N.T.; Lok, K.S.; Lee, P.P.; Su, M.; Wu, M.; Kocgozlu, L.; Ladoux, B. Multiarray cell stretching platform for high-magnification real-time imaging. Nanomedicine 2013, 8, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Kim, H.J.; Fraser, J.P.; Shea, D.E.; Khan, M.; Bahinski, A.; Hamilton, G.A.; Ingber, D.E. Microfabrication of human organs-on-chips. Nat. Protoc. 2013, 8, 2135–2157. [Google Scholar] [CrossRef] [PubMed]
- Benam, K.H.; Dauth, S.; Hassell, B.; Herland, A.; Jain, A.; Jang, K.J.; Karalis, K.; Kim, H.J.; MacQueen, L.; Mahmoodian, R.; et al. Engineered in vitro disease models. Annu. Rev. Pathol. 2015, 10, 195–262. [Google Scholar] [CrossRef] [PubMed]
- Avdeef, A.; Tam, K.Y. How well can the Caco-2/madin-darby canine kidney models predict effective human jejunal permeability? J. Med. Chem. 2010, 53, 3566–3584. [Google Scholar] [CrossRef] [PubMed]
- Bures, J.; Cyrany, J.; Kohoutova, D.; Forstl, M.; Rejchrt, S.; Kvetina, J.; Vorisek, V.; Kopacova, M. Small intestinal bacterial overgrowth syndrome. World J. Gastroenterol. 2010, 16, 2978–2990. [Google Scholar] [CrossRef] [PubMed]
- Furrie, E.; Macfarlane, S.; Kennedy, A.; Cummings, J.H.; Walsh, S.V.; O’Neil D, A.; Macfarlane, G.T. Synbiotic therapy (Bifidobacterium longum/Synergy 1) initiates resolution of inflammation in patients with active ulcerative colitis: A randomised controlled pilot trial. Gut 2005, 54, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Kruis, W.; Fric, P.; Pokrotnieks, J.; Lukas, M.; Fixa, B.; Kascak, M.; Kamm, M.A.; Weismueller, J.; Beglinger, C.; Stolte, M.; et al. Maintaining remission of ulcerative colitis with the probiotic Escherichia coli nissle 1917 is as effective as with standard mesalazine. Gut 2004, 53, 1617–1623. [Google Scholar] [CrossRef] [PubMed]
- Scaldaferri, F.; Gerardi, V.; Lopetuso, L.R.; Del Zompo, F.; Mangiola, F.; Boskoski, I.; Bruno, G.; Petito, V.; Laterza, L.; Cammarota, G.; et al. Gut microbial flora, prebiotics, and probiotics in IBD: Their current usage and utility. Biomed. Res. Int. 2013, 2013, 435268. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.M.; Kelly, C.P.; Farraye, F.A. Clostridium difficile infection in the inflammatory bowel disease patient. Inflamm. Bowel Dis. 2013, 19, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, Y.; Bamba, H.; Okui, M.; Tamura, K.; Tanida, N.; Satomi, M.; Shimoyama, T.; Nishigami, T. Helicobacter pylori infection increases mucosal permeability of the stomach and intestine. Digestion 2001, 63, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Krakauer, T. Stimulant-dependent modulation of cytokines and chemokines by airway epithelial cells: Cross talk between pulmonary epithelial and peripheral blood mononuclear cells. Clin. Diagn. Lab. Immunol. 2002, 9, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Haller, D.; Holt, L.; Parlesak, A.; Zanga, J.; Bauerlein, A.; Sartor, R.B.; Jobin, C. Differential effect of immune cells on non-pathogenic gram-negative bacteria-induced nuclear factor-κb activation and pro-inflammatory gene expression in intestinal epithelial cells. Immunology 2004, 112, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.I. Host-microbe interaction: Inflammation for growth. Nature 2010, 467, 410–411. [Google Scholar] [CrossRef] [PubMed]
- Victor, D.W.; Quigley, E.M. Microbial therapy in liver disease: Probiotics probe the microbiome-gut-liver-brain axis. Gastroenterology 2014, 147, 1216–1218. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.A.; McVey Neufeld, K.A. Gut-brain axis: How the microbiome influences anxiety and depression. Trends Neurosci. 2013, 36, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Imura, Y.; Sato, K.; Yoshimura, E. Micro total bioassay system for ingested substances: Assessment of intestinal absorption, hepatic metabolism, and bioactivity. Anal. Chem. 2010, 82, 9983–9988. [Google Scholar] [CrossRef] [PubMed]
- Imura, Y.; Yoshimura, E.; Sato, K. Micro total bioassay system for oral drugs: Evaluation of gastrointestinal degradation, intestinal absorption, hepatic metabolism, and bioactivity. Anal. Sci. 2012, 28, 197–199. [Google Scholar] [CrossRef] [PubMed]
- Prot, J.M.; Maciel, L.; Bricks, T.; Merlier, F.; Cotton, J.; Paullier, P.; Bois, F.Y.; Leclerc, E. First pass intestinal and liver metabolism of paracetamol in a microfluidic platform coupled with a mathematical modeling as a means of evaluating adme processes in humans. Biotechnol. Bioeng. 2014, 111, 2027–2040. [Google Scholar] [CrossRef] [PubMed]
- Konishi, S.; Fujita, T.; Hattor, K.; Kono, Y.; Matsushit, Y. An openable artificial intestinal tract system for the in vitro evaluation of medicines. Microsyst. Nanoeng. 2015, 1, 15015. [Google Scholar] [CrossRef]
- Li, J.; Zhu, Z. Research and development of next generation of antibody-based therapeutics. Acta Pharmacol. Sin. 2010, 31, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Denmark, V.K.; Mayer, L. Current status of monoclonal antibody therapy for the treatment of inflammatory bowel disease: An update. Expert Rev. Clin. Immunol. 2013, 9, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Fournier, B.M.; Parkos, C.A. The role of neutrophils during intestinal inflammation. Mucosal Immunol. 2012, 5, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Shah, U.; Dickinson, B.L.; Blumberg, R.S.; Simister, N.E.; Lencer, W.I.; Walker, W.A. Distribution of the IgG Fc receptor, FcRn, in the human fetal intestine. Pediatr. Res. 2003, 53, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Israel, E.J.; Taylor, S.; Wu, Z.; Mizoguchi, E.; Blumberg, R.S.; Bhan, A.; Simister, N.E. Expression of the neonatal Fc receptor, FcRn, on human intestinal epithelial cells. Immunology 1997, 92, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, B.L.; Badizadegan, K.; Wu, Z.; Ahouse, J.C.; Zhu, X.; Simister, N.E.; Blumberg, R.S.; Lencer, W.I. Bidirectional FcRn-dependent IgG transport in a polarized human intestinal epithelial cell line. J. Clin. Investig. 1999, 104, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Hornby, P.J.; Cooper, P.R.; Kliwinski, C.; Ragwan, E.; Mabus, J.R.; Harman, B.; Thompson, S.; Kauffman, A.L.; Yan, Z.; Tam, S.H.; et al. Human and non-human primate intestinal FcRn expression and immunoglobulin G transcytosis. Pharm. Res. 2014, 31, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Claypool, S.M.; Wagner, J.S.; Mizoguchi, E.; Mizoguchi, A.; Roopenian, D.C.; Lencer, W.I.; Blumberg, R.S. Human neonatal Fc receptor mediates transport of IgG into luminal secretions for delivery of antigens to mucosal dendritic cells. Immunity 2004, 20, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Masuda, A.; Kuo, T.T.; Kobayashi, K.; Claypool, S.M.; Takagawa, T.; Kutsumi, H.; Azuma, T.; Lencer, W.I.; Blumberg, R.S. IgG transport across mucosal barriers by neonatal Fc receptor for IgG and mucosal immunity. Springer Semin. Immunopathol. 2006, 28, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.L.; Chaudhury, C.; Kim, J.; Bronson, C.L.; Wani, M.A.; Mohanty, S. Perspective—FcRn transports albumin: Relevance to immunology and medicine. Trends Immunol. 2006, 27, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Suez, J.; Korem, T.; Zeevi, D.; Zilberman-Schapira, G.; Thaiss, C.A.; Maza, O.; Israeli, D.; Zmora, N.; Gilad, S.; Weinberger, A.; et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature 2014, 514, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, L.; McFall-Ngai, M.; Relman, D.A. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 2007, 449, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Mugambi, M.N.; Young, T.; Blaauw, R. Application of evidence on probiotics, prebiotics and synbiotics by food industry: A descriptive study. BMC Res. Notes 2014, 7, 754. [Google Scholar] [CrossRef] [PubMed]
- Frei, R.; Akdis, M.; O’Mahony, L. Prebiotics, probiotics, synbiotics, and the immune system: Experimental data and clinical evidence. Curr. Opin. Gastroenterol. 2015, 31, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, T.; Kojima, Y.; Seneviratne, C.J.; Maeda, N. Therapeutic application of synbiotics, a fusion of probiotics and prebiotics, and biogenics as a new concept for oral candida infections: A mini review. Front. Microbiol. 2016, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [PubMed]

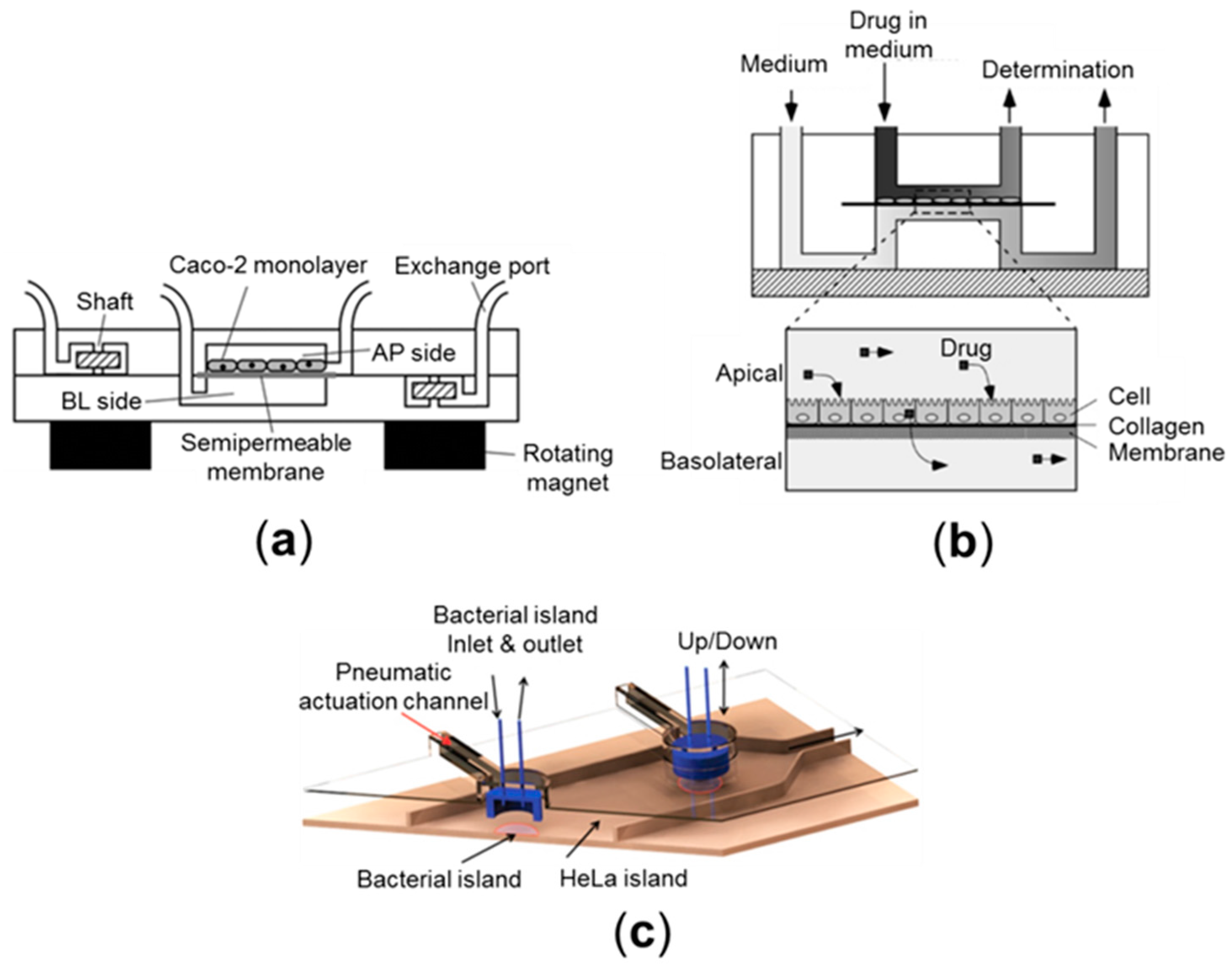




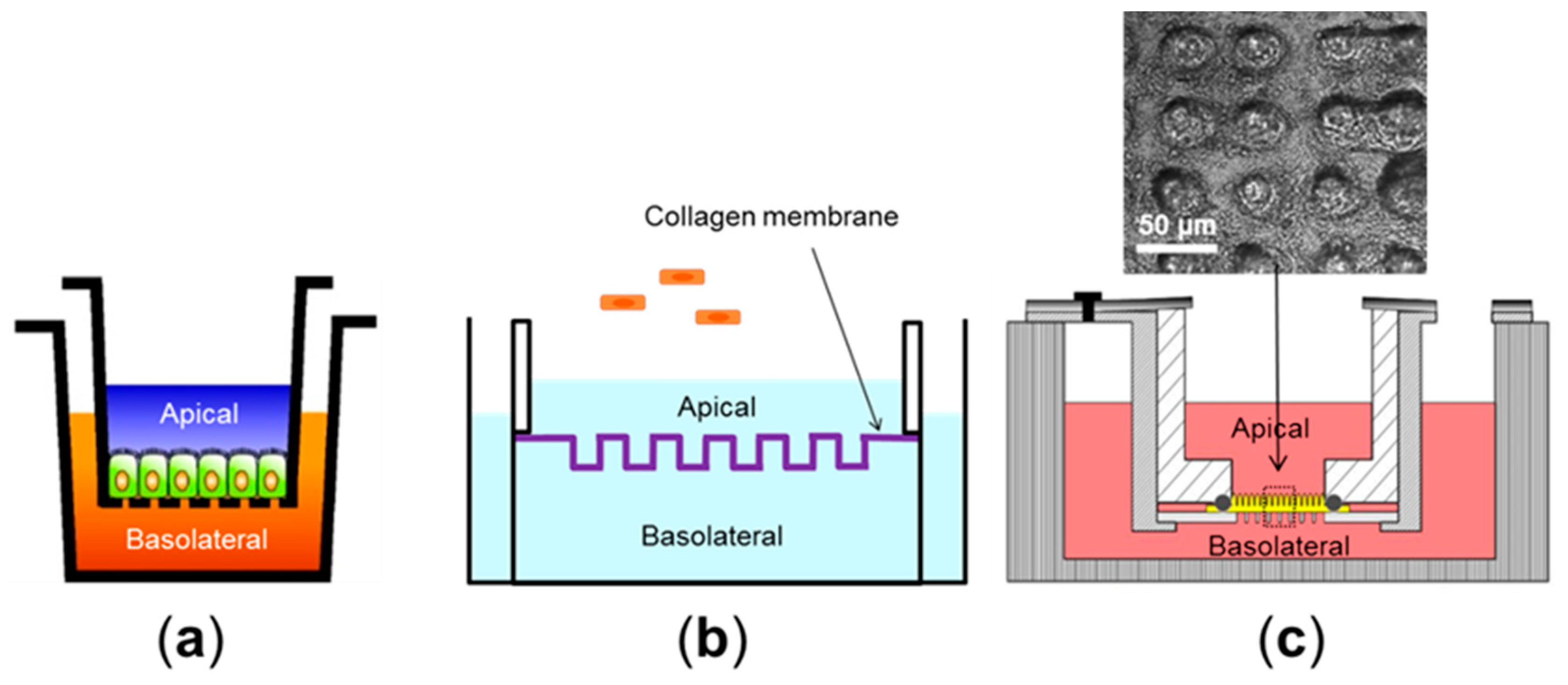
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Trade Name | Application | Target | Type |
|---|---|---|---|---|
| Adalimumab | Humira | CD | TNF-α | Mab 1 |
| Certolizumab pegol | Cimzia | CD | TNF-α | Fab' 2 |
| Fontolizumab | HuZAF | CD | IFN-γ | Mab |
| Infliximab | Remicade | CD/UC | TNF-α | Mab |
| Natalizumab | Tysabri | CD | integrin α4 | Mab |
| Visilizumab | Nuvion | CD/UC | CD3 | Mab |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, T.H.; Kim, H.J. Farewell to Animal Testing: Innovations on Human Intestinal Microphysiological Systems. Micromachines 2016, 7, 107. https://doi.org/10.3390/mi7070107
Kang TH, Kim HJ. Farewell to Animal Testing: Innovations on Human Intestinal Microphysiological Systems. Micromachines. 2016; 7(7):107. https://doi.org/10.3390/mi7070107
Chicago/Turabian StyleKang, Tae Hyun, and Hyun Jung Kim. 2016. "Farewell to Animal Testing: Innovations on Human Intestinal Microphysiological Systems" Micromachines 7, no. 7: 107. https://doi.org/10.3390/mi7070107
APA StyleKang, T. H., & Kim, H. J. (2016). Farewell to Animal Testing: Innovations on Human Intestinal Microphysiological Systems. Micromachines, 7(7), 107. https://doi.org/10.3390/mi7070107