
Ultrahigh-Throughput Improvement and Discovery of Enzymes Using Droplet-Based Microfluidic Screening

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
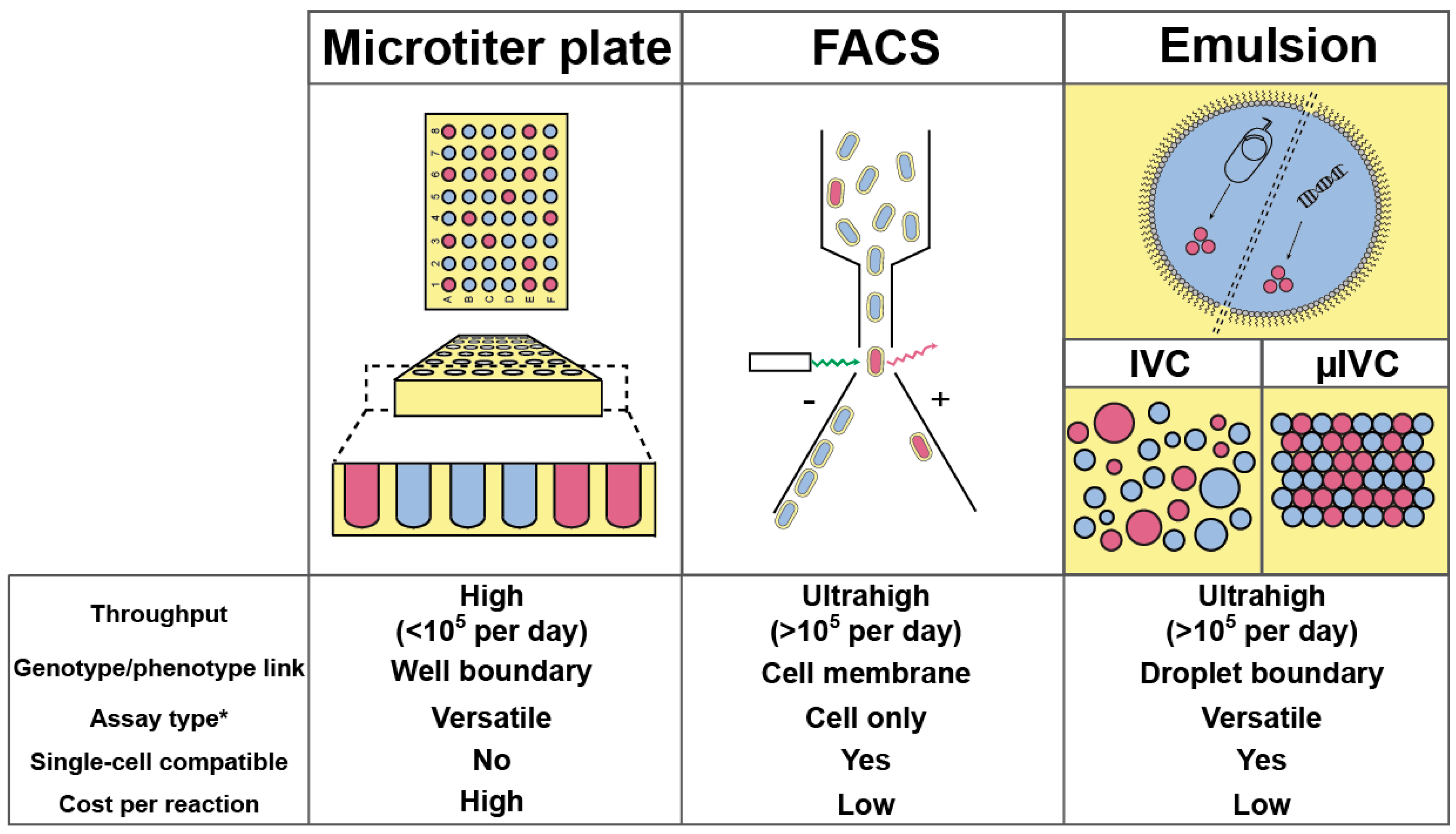
2. (Ultra) High-Throughput Screening Strategies and Their Limitations
3. Droplet-Based Microfluidics
3.1. Droplet Production
3.2. Droplet Content Modification
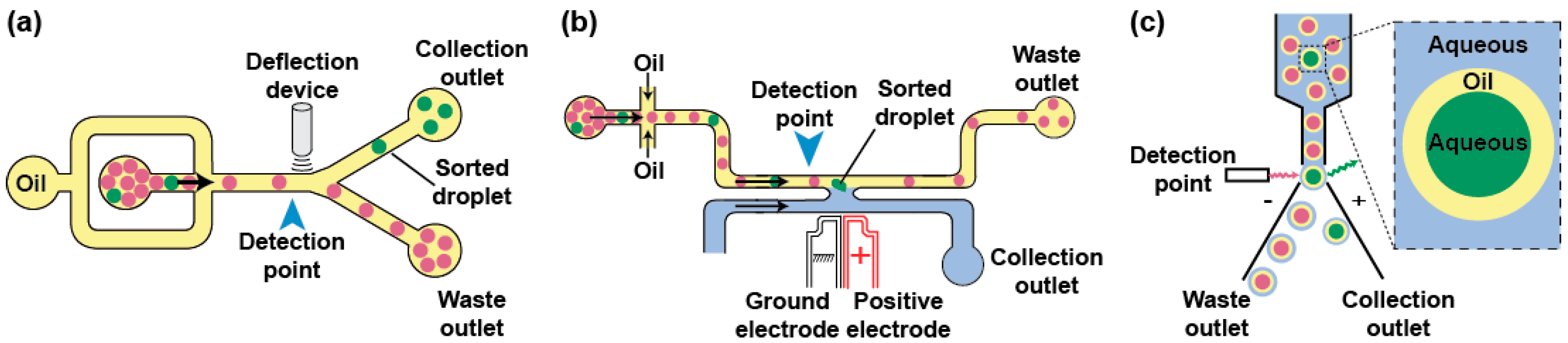
3.3. Droplet Analysis and Sorting
4. Genotype/Phenotype Confinement Maintenance
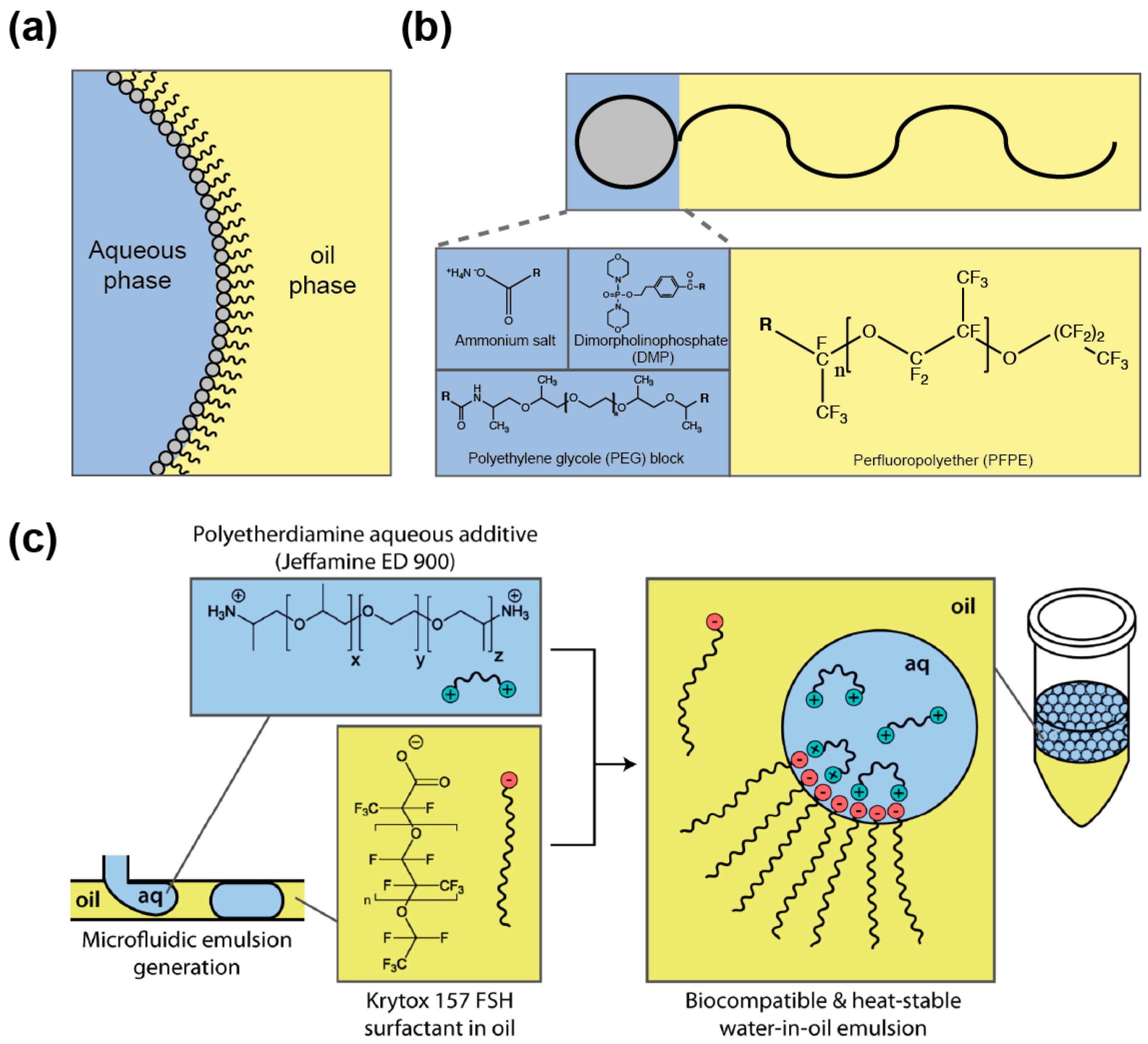
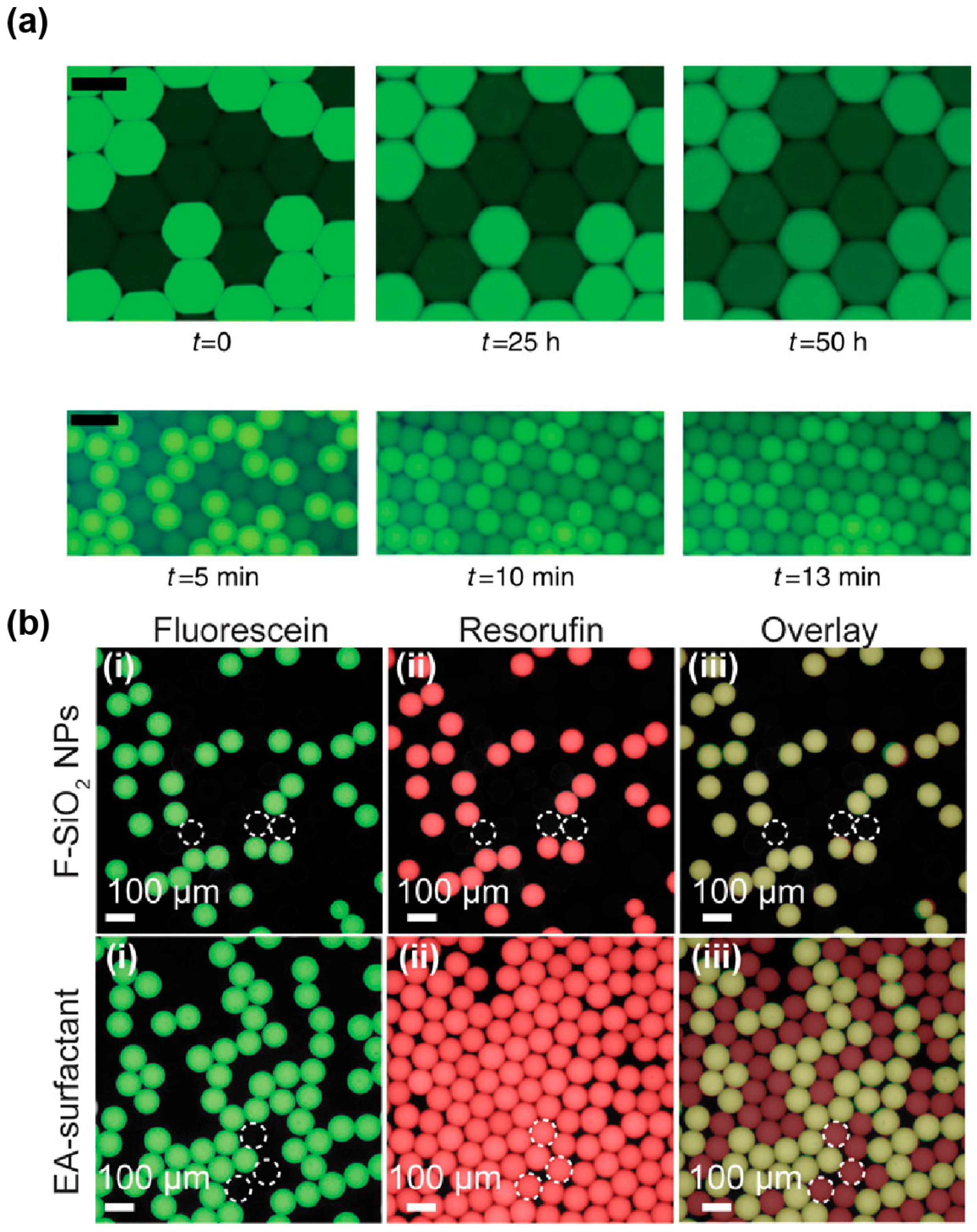
4.1. Droplet Stabilization
4.2. Limiting Droplet–Droplet Exchange
5. Discovery and Improvement of Biological Catalysts
5.1. Cell-Based Directed Evolution
5.2. Cell-Free Directed Evolution
5.3. Discovery of New Catalysts from Environmental Samples
6. Conclusions and Outlook
Acknowledgments
Conflicts of Interest
References
- Fersht, A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding; W.H. Freeman: New York, NY, USA, 1999; p. xxi. [Google Scholar]
- Jacques, P.; Bechet, M.; Bigan, M.; Caly, D.; Chataigne, G.; Coutte, F.; Flahaut, C.; Heuson, E.; Leclere, V.; Lecouturier, D.; et al. High-throughput strategies for the discovery and engineering of enzymes for biocatalysis. Bioprocess Biosyst. Eng. 2016, 40, 161–180. [Google Scholar] [CrossRef] [PubMed]
- Molina-Espeja, P.; Vina-Gonzalez, J.; Gomez-Fernandez, B.J.; Martin-Diaz, J.; Garcia-Ruiz, E.; Alcalde, M. Beyond the outer limits of nature by directed evolution. Biotechnol. Adv. 2016, 34, 754–767. [Google Scholar] [CrossRef] [PubMed]
- Khersonsky, O.; Tawfik, D.S. Enzyme promiscuity: A mechanistic and evolutionary perspective. Annu. Rev. Biochem. 2010, 79, 471–505. [Google Scholar] [PubMed]
- Pandya, C.; Farelli, J.D.; Dunaway-Mariano, D.; Allen, K.N. Enzyme promiscuity: Engine of evolutionary innovation. J. Biol. Chem. 2014, 289, 30229–30236. [Google Scholar] [CrossRef] [PubMed]
- Joyce, G.F. Forty years of in vitro evolution. Angew. Chem. 2007, 46, 6420–6436. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.A.; Arnold, F.H. Exploring protein fitness landscapes by directed evolution. Nat. Rev. Mol. Cell Biol. 2009, 10, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, M.; Tawfik, D.S. Directed enzyme evolution: Beyond the low-hanging fruit. Curr. Opin. Struct. Biol. 2012, 22, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Denard, C.A.; Ren, H.; Zhao, H. Improving and repurposing biocatalysts via directed evolution. Curr. Opin. Struct. Biol. 2015, 25, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Chockalingam, K.; Chen, Z. Directed evolution of enzymes and pathways for industrial biocatalysis. Curr. Opin. Biotechnol. 2002, 13, 104–110. [Google Scholar] [CrossRef]
- Wang, M.; Si, T.; Zhao, H. Biocatalyst development by directed evolution. Bioresour. Technol. 2012, 115, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Bao, Z.; Zhao, H. High throughput screening and selection methods for directed enzyme evolution. Ind. Eng. Chem. Res. 2015, 54, 4011–4020. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.L.; Rusli, R.A.; Ollis, D.L. Directed evolution of enzymes for industrial biocatalysis. Chembiochem Eur. J. Chem. Biol. 2016, 17, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, S. Directed evolution: Tailoring biocatalysts for industrial applications. Crit. Rev. Biotechnol. 2013, 33, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Gacio, A.; Uguen, M.; Fastrez, J. Phage display as a tool for the directed evolution of enzymes. Trends Biotechnol. 2003, 21, 408–414. [Google Scholar] [CrossRef]
- Guo, M.T.; Rotem, A.; Heyman, J.A.; Weitz, D.A. Droplet microfluidics for high-throughput biological assays. Lab Chip 2012, 12, 2146–2155. [Google Scholar] [CrossRef] [PubMed]
- Price, A.K.; Paegel, B.M. Discovery in droplets. Anal. Chem. 2016, 88, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Rakszewska, A.; Tel, J.; Chokkalingam, V.; Huck, W.T.S. One drop at a time: Toward droplet microfluidics as a versatile tool for single-cell analysis. NPG Asia Mater. 2014, 6, e133. [Google Scholar] [CrossRef]
- Shembekar, N.; Chaipan, C.; Utharala, R.; Merten, C.A. Droplet-based microfluidics in drug discovery, transcriptomics and high-throughput molecular genetics. Lab Chip 2016, 16, 1314–1331. [Google Scholar] [CrossRef] [PubMed]
- Mayr, L.M.; Bojanic, D. Novel trends in high-throughput screening. Curr. Opin. Pharmacol. 2009, 9, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Withers, S.G. Ultrahigh-throughput FACS-based screening for directed enzyme evolution. Chembiochem Eur. J. Chem. Biol. 2009, 10, 2704–2715. [Google Scholar] [CrossRef] [PubMed]
- Colin, P.Y.; Zinchenko, A.; Hollfelder, F. Enzyme engineering in biomimetic compartments. Curr. Opin. Struct. Biol. 2015, 33, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, D.S.; Griffiths, A.D. Man-made cell-like compartments for molecular evolution. Nat. Biotechnol. 1998, 16, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, A.D.; Tawfik, D.S. Miniaturising the laboratory in emulsion droplets. Trends Biotechnol. 2006, 24, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.T.; Baret, J.C.; Taly, V.; Griffiths, A.D. Miniaturizing chemistry and biology in microdroplets. Chem. Commun. 2007, 18, 1773–1788. [Google Scholar] [CrossRef] [PubMed]
- Bernath, K.; Hai, M.; Mastrobattista, E.; Griffiths, A.D.; Magdassi, S.; Tawfik, D.S. In vitro compartmentalization by double emulsions: Sorting and gene enrichment by fluorescence activated cell sorting. Anal. Biochem. 2004, 325, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, A.; Amitai, G.; Bernath, K.; Magdassi, S.; Tawfik, D.S. High-throughput screening of enzyme libraries: Thiolactonases evolved by fluorescence-activated sorting of single cells in emulsion compartments. Chem. Biol. 2005, 12, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.D.; Goldsmith, M.; Ashani, Y.; Simo, Y.; Mullokandov, G.; Bar, H.; Ben-David, M.; Leader, H.; Margalit, R.; Silman, I.; et al. Directed evolution of hydrolases for prevention of g-type nerve agent intoxication. Nat. Chem. Biol. 2011, 7, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, E.; Gibbs, M.; Reeves, R.; Bergquist, P. Directed evolution of a thermophilic beta-glucosidase for cellulosic bioethanol production. Appl. Biochem. Biotechnol. 2010, 161, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Xie, Y.; Huang, C.; Feng, Y.; Yang, G. An improved single cell ultrahigh throughput screening method based on in vitro compartmentalization. PLoS ONE 2014, 9, e89785. [Google Scholar] [CrossRef] [PubMed]
- Mastrobattista, E.; Taly, V.; Chanudet, E.; Treacy, P.; Kelly, B.T.; Griffiths, A.D. High-throughput screening of enzyme libraries: In vitro evolution of a beta-galactosidase by fluorescence-activated sorting of double emulsions. Chem. Biol. 2005, 12, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Ostafe, R.; Prodanovic, R.; Commandeur, U.; Fischer, R. Flow cytometry-based ultra-high-throughput screening assay for cellulase activity. Anal. Biochem. 2013, 435, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Ostafe, R.; Prodanovic, R.; Nazor, J.; Fischer, R. Ultra-high-throughput screening method for the directed evolution of glucose oxidase. Chem. Biol. 2014, 21, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Tu, R.; Martinez, R.; Prodanovic, R.; Klein, M.; Schwaneberg, U. A flow cytometry-based screening system for directed evolution of proteases. J. Biomol. Screen. 2011, 16, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Ghadessy, F.J.; Ong, J.L.; Holliger, P. Directed evolution of polymerase function by compartmentalized self-replication. Proc. Natl. Acad. Sci. USA 2001, 98, 4552–4557. [Google Scholar] [CrossRef] [PubMed]
- Zaher, H.S.; Unrau, P.J. Selection of an improved rna polymerase ribozyme with superior extension and fidelity. RNA 2007, 13, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Griswold, K.E.; Ellington, A.D. Direct selection of trans-acting ligase ribozymes by in vitro compartmentalization. RNA 2005, 11, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Wochner, A.; Attwater, J.; Coulson, A.; Holliger, P. Ribozyme-catalyzed transcription of an active ribozyme. Science 2011, 332, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Fischer, M.; Han, Y.; Withers, S.G.; Feng, Y.; Yang, G.Y. Substrate engineering enabling fluorescence droplet entrapment for IVC-FACS-based ultrahigh-throughput screening. Anal. Chem. 2016, 88, 8587–8595. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Wang, L. Passive and active droplet generation with microfluidics: A review. Lab Chip 2016, 17, 34–75. [Google Scholar] [CrossRef] [PubMed]
- Umbanhowar, P.B.; Prasad, V.; Weitz, D.A. Monodisperse emulsion generation via drop break off in a coflowing stream. Langmuir 2000, 16, 347–351. [Google Scholar] [CrossRef]
- Thorsen, T.; Roberts, R.W.; Arnold, F.H.; Quake, S.R. Dynamic pattern formation in a vesicle-generating microfluidic device. Phys. Rev. Lett. 2001, 86, 4163–4166. [Google Scholar] [CrossRef] [PubMed]
- Garstecki, P.; Fuerstman, M.J.; Stone, H.A.; Whitesides, G.M. Formation of droplets and bubbles in a microfluidic T-junction—Scaling and mechanism of break-up. Lab Chip 2006, 6, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Anna, S.L.; Bontoux, N.; Stone, H.A. Formation of dispersions using “flow focusing” in microchannels. Appl. Phys. Lett. 2003, 82, 364–366. [Google Scholar] [CrossRef]
- Abate, A.R.; Poitzsch, A.; Hwang, Y.; Lee, J.; Czerwinska, J.; Weitz, D.A. Impact of inlet channel geometry on microfluidic drop formation. Phys. Rev. E 2009, 80, 026310. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.M.; Alivisatos, A.P.; Mathies, R.A. High-temperature microfluidic synthesis of CdSe nanocrystals in nanoliter droplets. J. Am. Chem. Soc. 2005, 127, 13854–13861. [Google Scholar] [CrossRef] [PubMed]
- Priest, C.; Herminghaus, S.; Seemann, R. Generation of monodisperse gel emulsions in a microfluidic device. Appl. Phys. Lett. 2006, 88, 024106. [Google Scholar] [CrossRef]
- Dangla, R.; Kayi, S.C.; Baroud, C.N. Droplet microfluidics driven by gradients of confinement. Proc. Natl. Acad. Sci. USA 2013, 110, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Malloggi, F.; Pannacci, N.; Attia, R.; Monti, F.; Mary, P.; Willaime, H.; Tabeling, P.; Cabane, B.; Poncet, P. Monodisperse colloids synthesized with nanofluidic technology. Langmuir 2010, 26, 2369–2373. [Google Scholar] [CrossRef] [PubMed]
- Rhee, M.; Burns, M.A. Drop mixing in a microchannel for lab-on-a-chip platforms. Langmuir 2008, 24, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Tice, J.D.; Ismagilov, R.F. A microfluidic system for controlling reaction networks in time. Angew. Chem. Int. Ed. 2003, 42, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Courtois, F.; Olguin, L.F.; Whyte, G.; Bratton, D.; Huck, W.T.; Abell, C.; Hollfelder, F. An integrated device for monitoring time-dependent in vitro expression from single genes in picolitre droplets. Chembiochem Eur. J. Chem. Biol. 2008, 9, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Khorshidi, M.A.; Rajeswari, P.K.P.; Wahlby, C.; Joensson, H.N.; Svahn, H.A. Automated analysis of dynamic behavior of single cells in picoliter droplets. Lab Chip 2014, 14, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Huebner, A.; Bratton, D.; Whyte, G.; Yang, M.; Demello, A.J.; Abell, C.; Hollfelder, F. Static microdroplet arrays: A microfluidic device for droplet trapping, incubation and release for enzymatic and cell-based assays. Lab Chip 2009, 9, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Ismagilov, R.F. Millisecond kinetics on a microfluidic chip using nanoliters of reagents. J. Am. Chem. Soc. 2003, 125, 14613–14619. [Google Scholar] [CrossRef] [PubMed]
- Frenz, L.; Blank, K.; Brouzes, E.; Griffiths, A.D. Reliable microfluidic on-chip incubation of droplets in delay-lines. Lab Chip 2009, 9, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Pekin, D.; Skhiri, Y.; Baret, J.C.; Le Corre, D.; Mazutis, L.; Salem, C.B.; Millot, F.; El Harrak, A.; Hutchison, J.B.; Larson, J.W.; et al. Quantitative and sensitive detection of rare mutations using droplet-based microfluidics. Lab Chip 2011, 11, 2156–2166. [Google Scholar] [CrossRef] [PubMed]
- Mazutis, L.; Araghi, A.F.; Miller, O.J.; Baret, J.C.; Frenz, L.; Janoshazi, A.; Taly, V.; Miller, B.J.; Hutchison, J.B.; Link, D.; et al. Droplet-based microfluidic systems for high-throughput single DNA molecule isothermal amplification and analysis. Anal. Chem. 2009, 81, 4813–4821. [Google Scholar] [CrossRef] [PubMed]
- Baret, J.C.; Miller, O.J.; Taly, V.; Ryckelynck, M.; El-Harrak, A.; Frenz, L.; Rick, C.; Samuels, M.L.; Hutchison, J.B.; Agresti, J.J.; et al. Fluorescence-activated droplet sorting (FADS): Efficient microfluidic cell sorting based on enzymatic activity. Lab Chip 2009, 9, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Kintses, B.; Hein, C.; Mohamed, M.F.; Fischlechner, M.; Courtois, F.; Laine, C.; Hollfelder, F. Picoliter cell lysate assays in microfluidic droplet compartments for directed enzyme evolution. Chem. Biol. 2012, 19, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Li, Q.; Kennedy, R.T. Rapid and label-free screening of enzyme inhibitors using segmented flow electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 2010, 21, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Kennedy, R.T. Droplet electrospray ionization mass spectrometry for high throughput screening for enzyme inhibitors. Anal. Chem. 2014, 86, 9309–9314. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Fang, Q. High-throughput sample introduction for droplet-based screening with an on-chip integrated sampling probe and slotted-vial array. Lab Chip 2010, 10, 2864–2868. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.F.; Zhu, Y.; Du, G.S.; Fang, Q. Droplet-based microfluidic flow injection system with large-scale concentration gradient by a single nanoliter-scale injection for enzyme inhibition assay. Anal. Chem. 2012, 84, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Gielen, F.; van Vliet, L.; Koprowski, B.T.; Devenish, S.R.; Fischlechner, M.; Edel, J.B.; Niu, X.; deMello, A.J.; Hollfelder, F. A fully unsupervised compartment-on-demand platform for precise nanoliter assays of time-dependent steady-state enzyme kinetics and inhibition. Anal. Chem. 2013, 85, 4761–4769. [Google Scholar] [CrossRef] [PubMed]
- Price, A.K.; MacConnell, A.B.; Paegel, B.M. Hnusabr: Photochemical dose-response bead screening in droplets. Anal. Chem. 2016, 88, 2904–2911. [Google Scholar] [CrossRef] [PubMed]
- Jambovane, S.; Kim, D.J.; Duin, E.C.; Kim, S.K.; Hong, J.W. Creation of stepwise concentration gradient in picoliter droplets for parallel reactions of matrix metalloproteinase ii and ix. Anal. Chem. 2011, 83, 3358–3364. [Google Scholar] [CrossRef] [PubMed]
- Rane, T.D.; Zec, H.C.; Wang, T.H. A barcode-free combinatorial screening platform for matrix metalloproteinase screening. Anal. Chem. 2015, 87, 1950–1956. [Google Scholar] [CrossRef] [PubMed]
- Toh, A.G.G.; Wang, Z.P.; Yang, C.; Nguyen, N.T. Engineering microfluidic concentration gradient generators for biological applications. Microfluid. Nanofluid. 2014, 16, 1–18. [Google Scholar] [CrossRef]
- Bui, M.P.N.; Li, C.A.; Han, K.N.; Choo, J.; Lee, E.K.; Seong, G.H. Enzyme kinetic measurements using a droplet-based microfluidic system with a concentration gradient. Anal. Chem. 2011, 83, 1603–1608. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Sarkar, A.; Song, Y.A.; Miller, M.A.; Kim, S.J.; Griffith, L.G.; Lauffenburger, D.A.; Han, J. Enhancing protease activity assay in droplet-based microfluidics using a biomolecule concentrator. J. Am. Chem. Soc. 2011, 133, 10368–10371. [Google Scholar] [CrossRef] [PubMed]
- Theberge, A.B.; Whyte, G.; Huck, W.T. Generation of picoliter droplets with defined contents and concentration gradients from the separation of chemical mixtures. Anal. Chem. 2010, 82, 3449–3453. [Google Scholar] [CrossRef] [PubMed]
- Beneyton, T.; Coldren, F.; Baret, J.C.; Griffiths, A.D.; Taly, V. Cota laccase: High-throughput manipulation and analysis of recombinant enzyme libraries expressed in E. coli using droplet-based microfluidics. Analyst 2014, 139, 3314–3323. [Google Scholar] [CrossRef] [PubMed]
- Hammar, P.; Angermayr, S.A.; Sjostrom, S.L.; van der Meer, J.; Hellingwerf, K.J.; Hudson, E.P.; Joensson, H.N. Single-cell screening of photosynthetic growth and lactate production by cyanobacteria. Biotechnol. Biofuels 2015, 8, 193. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.L.; Ghaderi, A.; Zhou, H.; Agresti, J.; Weitz, D.A.; Fink, G.R.; Stephanopoulos, G. Microfluidic high-throughput culturing of single cells for selection based on extracellular metabolite production or consumption. Nat. Biotechnol. 2014, 32, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Mazutis, L.; Baret, J.C.; Treacy, P.; Skhiri, Y.; Araghi, A.F.; Ryckelynck, M.; Taly, V.; Griffiths, A.D. Multi-step microfluidic droplet processing: Kinetic analysis of an in vitro translated enzyme. Lab Chip 2009, 9, 2902–2908. [Google Scholar] [CrossRef] [PubMed]
- Baroud, C.N.; Gallaire, F.; Dangla, R. Dynamics of microfluidic droplets. Lab Chip 2010, 10, 2032–2045. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Gulati, S.; Edel, J.B.; deMello, A.J. Pillar-induced droplet merging in microfluidic circuits. Lab Chip 2008, 8, 1837–1841. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; Agresti, J.; Chong, H.; Marquez, M.; Weitz, D.A. Electrocoalescence of drops synchronized by size-dependent flow in microfluidic channels. Appl. Phys. Lett. 2006, 88, 264105. [Google Scholar] [CrossRef]
- El Debs, B.; Utharala, R.; Balyasnikova, I.V.; Griffiths, A.D.; Merten, C.A. Functional single-cell hybridoma screening using droplet-based microfluidics. Proc. Natl. Acad. Sci. USA 2012, 109, 11570–11575. [Google Scholar] [CrossRef] [PubMed]
- Mazutis, L.; Griffiths, A.D. Selective droplet coalescence using microfluidic systems. Lab Chip 2012, 12, 1800–1806. [Google Scholar] [CrossRef] [PubMed]
- Bremond, N.; Thiam, A.R.; Bibette, J. Decompressing emulsion droplets favors coalescence. Phys. Rev. lett. 2008, 100, 024501. [Google Scholar] [CrossRef] [PubMed]
- Baroud, C.N.; de Saint Vincent, M.R.; Delville, J.P. An optical toolbox for total control of droplet microfluidics. Lab Chip 2007, 7, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Link, D.R.; Grasland-Mongrain, E.; Duri, A.; Sarrazin, F.; Cheng, Z.; Cristobal, G.; Marquez, M.; Weitz, D.A. Electric control of droplets in microfluidic devices. Angew. Chem. Int. Ed. 2006, 45, 2556–2560. [Google Scholar] [CrossRef] [PubMed]
- Chabert, M.; Dorfman, K.D.; Viovy, J.L. Droplet fusion by alternating current (AC) field electrocoalescence in microchannels. Electrophoresis 2005, 26, 3706–3715. [Google Scholar] [CrossRef] [PubMed]
- Priest, C.; Herminghaus, S.; Seemann, R. Controlled electrocoalescence in microfluidics: Targeting a single lamella. Appl. Phys. Lett. 2006, 89, 134101. [Google Scholar] [CrossRef]
- Frenz, L.; El Harrak, A.; Pauly, M.; Begin-Colin, S.; Griffiths, A.D.; Baret, J.C. Droplet-based microreactors for the synthesis of magnetic iron oxide nanoparticles. Angew. Chem. Int. Ed. 2008, 47, 6817–6820. [Google Scholar] [CrossRef] [PubMed]
- Sciambi, A.; Abate, A.R. Generating electric fields in PDMS microfluidic devices with salt water electrodes. Lab Chip 2014, 14, 2605–2609. [Google Scholar] [CrossRef] [PubMed]
- Abate, A.R.; Hung, T.; Mary, P.; Agresti, J.J.; Weitz, D.A. High-throughput injection with microfluidics using picoinjectors. Proc. Natl. Acad. Sci. USA 2010, 107, 19163–19166. [Google Scholar] [CrossRef] [PubMed]
- Rhee, M.; Light, Y.K.; Yilmaz, S.; Adams, P.D.; Saxena, D.; Meagher, R.J.; Singh, A.K. Pressure stabilizer for reproducible picoinjection in droplet microfluidic systems. Lab Chip 2014, 14, 4533–4539. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Fang, Q. Analytical detection techniques for droplet microfluidics—A review. Anal. Chim. Acta 2013, 787, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Hess, D.; Rane, A.; deMello, A.J.; Stavrakis, S. High-throughput, quantitative enzyme kinetic analysis in microdroplets using stroboscopic epifluorescence imaging. Anal. Chem. 2015, 87, 4965–4972. [Google Scholar] [CrossRef] [PubMed]
- Deal, K.S.; Easley, C.J. Self-regulated, droplet-based sample chopper for microfluidic absorbance detection. Anal. Chem. 2012, 84, 1510–1516. [Google Scholar] [CrossRef] [PubMed]
- Gielen, F.; Hours, R.; Emond, S.; Fischlechner, M.; Schell, U.; Hollfelder, F. Ultrahigh-throughput-directed enzyme evolution by absorbance-activated droplet sorting (AADS). Proc. Natl. Acad. Sci. USA 2016, 113, E7383–E7389. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.D.; Zheng, H.; Guo, W.; Ganan-Calvo, A.M.; Ai, Y.; Tsao, C.W.; Zhou, J.; Li, W.; Huang, Y.; Nguyen, N.T.; et al. Active droplet sorting in microfluidics: A review. Lab Chip 2017, 17, 751–771. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, J.; Bransky, A.; Khoury, M.; Levenberg, S. Advanced microfluidic droplet manipulation based on piezoelectric actuation. Biomed. Microdevices 2010, 12, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Abate, A.R.; Agresti, J.J.; Weitz, D.A. Microfluidic sorting with high-speed single-layer membrane valves. Appl. Phys. Lett. 2010, 96, 203509. [Google Scholar] [CrossRef]
- Franke, T.; Abate, A.R.; Weitz, D.A.; Wixforth, A. Surface acoustic wave (SAW) directed droplet flow in microfluidics for PDMS devices. Lab Chip 2009, 9, 2625–2627. [Google Scholar] [CrossRef] [PubMed]
- Schmid, L.; Weitz, D.A.; Franke, T. Sorting drops and cells with acoustics: Acoustic microfluidic fluorescence-activated cell sorter. Lab Chip 2014, 14, 3710–3718. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.; Lee, K.; Panchapakesan, R.; Oh, K.W. On-demand electrostatic droplet charging and sorting. Biomicrofluidics 2011, 5, 024113. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; Kerbage, C.; Hunt, T.P.; Westervelt, R.M.; Link, D.R.; Weitz, D.A. Dielectrophoretic manipulation of drops for high-speed microfluidic sorting devices. Appl. Phys. Lett. 2006, 88, 024104. [Google Scholar] [CrossRef]
- Sciambi, A.; Abate, A.R. Accurate microfluidic sorting of droplets at 30 kHz. Lab Chip 2015, 15, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Fidalgo, L.M.; Whyte, G.; Bratton, D.; Kaminski, C.F.; Abell, C.; Huck, W.T. From microdroplets to microfluidics: Selective emulsion separation in microfluidic devices. Angew. Chem. Int. Ed. 2008, 47, 2042–2045. [Google Scholar] [CrossRef] [PubMed]
- Fallah-Araghi, A.; Baret, J.C.; Ryckelynck, M.; Griffiths, A.D. A completely in vitro ultrahigh-throughput droplet-based microfluidic screening system for protein engineering and directed evolution. Lab Chip 2012, 12, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.A.; Tran, T.M.; Abate, A.R. Dissecting enzyme function with microfluidic-based deep mutational scanning. Proc. Natl. Acad. Sci. USA 2015, 112, 7159–7164. [Google Scholar] [CrossRef] [PubMed]
- Zinchenko, A.; Devenish, S.R.; Kintses, B.; Colin, P.Y.; Fischlechner, M.; Hollfelder, F. One in a million: Flow cytometric sorting of single cell-lysate assays in monodisperse picolitre double emulsion droplets for directed evolution. Anal. Chem. 2014, 86, 2526–2533. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Yang, C.J. Hydrogel droplet microfluidics for high-throughput single molecule/cell analysis. Acc. Chem. Res. 2016, 50, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Fischlechner, M.; Schaerli, Y.; Mohamed, M.F.; Patil, S.; Abell, C.; Hollfelder, F. Evolution of enzyme catalysts caged in biomimetic gel-shell beads. Nat. Chem. 2014, 6, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Baret, J.C. Surfactants in droplet-based microfluidics. Lab Chip 2012, 12, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Gruner, P.; Riechers, B.; Orellana, L.A.C.; Brosseau, Q.; Maes, F.; Beneyton, T.; Pekin, D.; Baret, J.C. Stabilisers for water-in-fluorinated-oil dispersions: Key properties for microfluidic applications. Curr. Opin. Colloid Interface Sci. 2015, 20, 183–191. [Google Scholar] [CrossRef]
- Miller, O.J.; Bernath, K.; Agresti, J.J.; Amitai, G.; Kelly, B.T.; Mastrobattista, E.; Taly, V.; Magdassi, S.; Tawfik, D.S.; Griffiths, A.D. Directed evolution by in vitro compartmentalization. Nat. Methods 2006, 3, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Studer, A.; Hadida, S.; Ferritto, R.; Kim, S.Y.; Jeger, P.; Wipf, P.; Curran, D.P. Fluorous synthesis: A fluorous-phase strategy for improving separation efficiency in organic synthesis. Science 1997, 275, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.N.; Park, C.; Whitesides, G.M. Solvent compatibility of poly(dimethylsiloxane)-based microfluidic devices. Anal. Chem. 2003, 75, 6544–6554. [Google Scholar] [CrossRef] [PubMed]
- Lowe, K.C.; Anthony, P.; Wardrop, J.; Davey, M.R.; Power, J.B. Perfluorochemicals and cell biotechnology. Artif. Cell Blood Substit. Immobil. Biotechnol. 1997, 25, 261–274. [Google Scholar] [CrossRef]
- Clausell-Tormos, J.; Lieber, D.; Baret, J.C.; El-Harrak, A.; Miller, O.J.; Frenz, L.; Blouwolff, J.; Humphry, K.J.; Koster, S.; Duan, H.; et al. Droplet-based microfluidic platforms for the encapsulation and screening of mammalian cells and multicellular organisms. Chem. Biol. 2008, 15, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Kaltenbach, M.; Devenish, S.R.; Hollfelder, F. A simple method to evaluate the biochemical compatibility of oil/surfactant mixtures for experiments in microdroplets. Lab Chip 2012, 12, 4185–4192. [Google Scholar] [CrossRef] [PubMed]
- DeJournette, C.J.; Kim, J.; Medlen, H.; Li, X.; Vincent, L.J.; Easley, C.J. Creating biocompatible oil-water interfaces without synthesis: Direct interactions between primary amines and carboxylated perfluorocarbon surfactants. Anal. Chem. 2013, 85, 10556–10564. [Google Scholar] [CrossRef] [PubMed]
- Holtze, C.; Rowat, A.C.; Agresti, J.J.; Hutchison, J.B.; Angile, F.E.; Schmitz, C.H.J.; Koster, S.; Duan, H.; Humphry, K.J.; Scanga, R.A.; et al. Biocompatible surfactants for water-in-fluorocarbon emulsions. Lab Chip 2008, 8, 1632–1639. [Google Scholar] [CrossRef] [PubMed]
- Brouzes, E.; Medkova, M.; Savenelli, N.; Marran, D.; Twardowski, M.; Hutchison, J.B.; Rothberg, J.M.; Link, D.R.; Perrimon, N.; Samuels, M.L. Droplet microfluidic technology for single-cell high-throughput screening. Proc. Natl. Acad. Sci. USA 2009, 106, 14195–14200. [Google Scholar] [CrossRef] [PubMed]
- Kreutz, J.E.; Li, L.; Roach, L.S.; Hatakeyama, T.; Ismagilov, R.F. Laterally mobile, functionalized self-assembled monolayers at the fluorous-aqueous interface in a plug-based microfluidic system: Characterization and testing with membrane protein crystallization. J. Am. Chem. Soc. 2009, 131, 6042–6043. [Google Scholar] [CrossRef] [PubMed]
- Platzman, I.; Janiesch, J.W.; Spatz, J.P. Synthesis of nanostructured and biofunctionalized water-in-oil droplets as tools for homing T cells. J. Am. Chem. Soc. 2013, 135, 3339–3342. [Google Scholar] [CrossRef] [PubMed]
- Wagner, O.; Thiele, J.; Weinhart, M.; Mazutis, L.; Weitz, D.A.; Huck, W.T.; Haag, R. Biocompatible fluorinated polyglycerols for droplet microfluidics as an alternative to peg-based copolymer surfactants. Lab Chip 2016, 16, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Ursuegui, S.; Mosser, M.; Wagner, A. Copper-free click chemistry for microdroplet‘s w/o interface engineering. RSC Adv. 2016, 6, 94942–94948. [Google Scholar] [CrossRef]
- Pays, K.; Giermanska-Kahn, J.; Pouligny, B.; Bibette, J.; Leal-Calderon, F. Double emulsions: How does release occur? J. Control. Release 2002, 79, 193–205. [Google Scholar] [CrossRef]
- Hai, M.; Magdassi, S. Investigation on the release of fluorescent markers from w/o/w emulsions by fluorescence-activated cell sorter. J. Control. Release 2004, 96, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Woronoff, G.; El Harrak, A.; Mayot, E.; Schicke, O.; Miller, O.J.; Soumillion, P.; Griffiths, A.D.; Ryckelynck, M. New generation of amino coumarin methyl sulfonate-based fluorogenic substrates for amidase assays in droplet-based microfluidic applications. Anal. Chem. 2011, 83, 2852–2857. [Google Scholar] [CrossRef] [PubMed]
- Skhiri, Y.; Gruner, P.; Semin, B.; Brosseau, Q.; Pekin, D.; Mazutis, L.; Goust, V.; Kleinschmidt, F.; El Harrak, A.; Hutchison, J.B.; et al. Dynamics of molecular transport by surfactants in emulsions. Soft Matter 2012, 8, 10618–10627. [Google Scholar] [CrossRef]
- Chen, Y.; Wijaya Gani, A.; Tang, S.K. Characterization of sensitivity and specificity in leaky droplet-based assays. Lab Chip 2012, 12, 5093–5103. [Google Scholar] [CrossRef] [PubMed]
- Gruner, P.; Riechers, B.; Semin, B.; Lim, J.; Johnston, A.; Short, K.; Baret, J.C. Controlling molecular transport in minimal emulsions. Nat. Commun. 2016, 7, 10392. [Google Scholar] [CrossRef] [PubMed]
- Janiesch, J.W.; Weiss, M.; Kannenberg, G.; Hannabuss, J.; Surrey, T.; Platzman, I.; Spatz, J.P. Key factors for stable retention of fluorophores and labeled biomolecules in droplet-based microfluidics. Anal. Chem. 2015, 87, 2063–2067. [Google Scholar] [CrossRef] [PubMed]
- Najah, M.; Mayot, E.; Mahendra-Wijaya, I.P.; Griffiths, A.D.; Ladame, S.; Drevelle, A. New glycosidase substrates for droplet-based microfluidic screening. Anal. Chem. 2013, 85, 9807–9814. [Google Scholar] [CrossRef] [PubMed]
- Scheler, O.; Kaminski, T.S.; Ruszczak, A.; Garstecki, P. Dodecylresorufin (C12R) outperforms resorufin in microdroplet bacterial assays. ACS Appl. Mater. Interfaces 2016, 8, 11318–11325. [Google Scholar] [CrossRef] [PubMed]
- Sandoz, P.A.; Chung, A.J.; Weaver, W.M.; Di Carlo, D. Sugar additives improve signal fidelity for implementing two-phase resorufin-based enzyme immunoassays. Langmuir 2014, 30, 6637–6643. [Google Scholar] [CrossRef] [PubMed]
- Courtois, F.; Olguin, L.F.; Whyte, G.; Theberge, A.B.; Huck, W.T.; Hollfelder, F.; Abell, C. Controlling the retention of small molecules in emulsion microdroplets for use in cell-based assays. Anal. Chem. 2009, 81, 3008–3016. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Rosenfeld, L.; Kim, M.; Xu, M.; Lin, E.; Derda, R.; Tang, S.K. Fluorinated pickering emulsions impede interfacial transport and form rigid interface for the growth of anchorage-dependent cells. ACS Appl. Mater. Interfaces 2014, 6, 21446–21453. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Lyu, F.; Tang, S.K. Fluorinated pickering emulsions with nonadsorbing interfaces for droplet-based enzymatic assays. Anal. Chem. 2015, 87, 7938–7943. [Google Scholar] [CrossRef] [PubMed]
- Agresti, J.J.; Antipov, E.; Abate, A.R.; Ahn, K.; Rowat, A.C.; Baret, J.C.; Marquez, M.; Klibanov, A.M.; Griffiths, A.D.; Weitz, D.A. Ultrahigh-throughput screening in drop-based microfluidics for directed evolution. Proc. Natl. Acad. Sci. USA 2010, 107, 4004–4009. [Google Scholar] [CrossRef] [PubMed]
- Huebner, A.; Srisa-Art, M.; Holt, D.; Abell, C.; Hollfelder, F.; deMello, A.J.; Edel, J.B. Quantitative detection of protein expression in single cells using droplet microfluidics. Chem. Commun. 2007, 1218–1220. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.C.; Dunn, M.R.; Hatch, A.; Sau, S.P.; Youngbull, C.; Chaput, J.C. A general strategy for expanding polymerase function by droplet microfluidics. Nat. Commun. 2016, 7, 11235. [Google Scholar] [CrossRef] [PubMed]
- Obexer, R.; Godina, A.; Garrabou, X.; Mittl, P.R.; Baker, D.; Griffiths, A.D.; Hilvert, D. Emergence of a catalytic tetrad during evolution of a highly active artificial aldolase. Nat. Chem. 2017, 9, 50–56. [Google Scholar] [CrossRef] [PubMed]
- De Lange, N.; Tran, T.M.; Abate, A.R. Electrical lysis of cells for detergent-free droplet assays. Biomicrofluidics 2016, 10, 024114. [Google Scholar] [CrossRef] [PubMed]
- Najah, M.; Griffiths, A.D.; Ryckelynck, M. Teaching single-cell digital analysis using droplet-based microfluidics. Anal. Chem. 2012, 84, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Ostafe, R.; Prodanovic, R.; Lloyd Ung, W.; Weitz, D.A.; Fischer, R. A high-throughput cellulase screening system based on droplet microfluidics. Biomicrofluidics 2014, 8, 041102. [Google Scholar] [CrossRef] [PubMed]
- Beneyton, T.; Wijaya, I.P.; Postros, P.; Najah, M.; Leblond, P.; Couvent, A.; Mayot, E.; Griffiths, A.D.; Drevelle, A. High-throughput screening of filamentous fungi using nanoliter-range droplet-based microfluidics. Sci. Rep. 2016, 6, 27223. [Google Scholar] [CrossRef] [PubMed]
- Sjostrom, S.L.; Bai, Y.; Huang, M.; Liu, Z.; Nielsen, J.; Joensson, H.N.; Andersson Svahn, H. High-throughput screening for industrial enzyme production hosts by droplet microfluidics. Lab Chip 2014, 14, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Bai, Y.; Sjostrom, S.L.; Hallstrom, B.M.; Liu, Z.; Petranovic, D.; Uhlen, M.; Joensson, H.N.; Andersson-Svahn, H.; Nielsen, J. Microfluidic screening and whole-genome sequencing identifies mutations associated with improved protein secretion by yeast. Proc. Natl. Acad. Sci. USA 2015, 112, E4689–E4696. [Google Scholar] [CrossRef] [PubMed]
- Colin, P.Y.; Kintses, B.; Gielen, F.; Miton, C.M.; Fischer, G.; Mohamed, M.F.; Hyvonen, M.; Morgavi, D.P.; Janssen, D.B.; Hollfelder, F. Ultrahigh-throughput discovery of promiscuous enzymes by picodroplet functional metagenomics. Nat. Commun. 2015, 6, 10008. [Google Scholar] [CrossRef] [PubMed]
- Ryckelynck, M.; Baudrey, S.; Rick, C.; Marin, A.; Coldren, F.; Westhof, E.; Griffiths, A.D. Using droplet-based microfluidics to improve the catalytic properties of rna under multiple-turnover conditions. RNA 2015, 21, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Inoue, A.; Tomari, Y.; Suzuki, T.; Yokogawa, T.; Nishikawa, K.; Ueda, T. Cell-free translation reconstituted with purified components. Nat Biotechnol. 2001, 19, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Dittrich, P.S.; Jahnz, M.; Schwille, P. A new embedded process for compartmentalized cell-free protein expression and on-line detection in microfluidic devices. Chembiochem Eur. J. Chem. Biol. 2005, 6, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Kiss, M.M.; Ortoleva-Donnelly, L.; Beer, N.R.; Warner, J.; Bailey, C.G.; Colston, B.W.; Rothberg, J.M.; Link, D.R.; Leamon, J.H. High-throughput quantitative polymerase chain reaction in picoliter droplets. Anal. Chem. 2008, 80, 8975–8981. [Google Scholar] [CrossRef] [PubMed]
- Woronoff, G.; Ryckelynck, M.; Wessel, J.; Schicke, O.; Griffiths, A.D.; Soumillion, P. Activity-fed translation (AFT) assay: A new high-throughput screening strategy for enzymes in droplets. Chembiochem Eur. J. Chem. Biol. 2015, 16, 1343–1349. [Google Scholar] [CrossRef] [PubMed]
- Schaerli, Y.; Wootton, R.C.; Robinson, T.; Stein, V.; Dunsby, C.; Neil, M.A.; French, P.M.; Demello, A.J.; Abell, C.; Hollfelder, F. Continuous-flow polymerase chain reaction of single-copy DNA in microfluidic microdroplets. Anal. Chem. 2009, 81, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Autour, A.; Westhof, E.; Ryckelynck, M. Ispinach: A fluorogenic RNA aptamer optimized for in vitro applications. Nucleic Acids Res. 2016, 44, 2491–2500. [Google Scholar] [CrossRef] [PubMed]
- Vartoukian, S.R.; Palmer, R.M.; Wade, W.G. Strategies for culture of ‘unculturable’ bacteria. FEMS Microbiol. Lett. 2010, 309, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ekkers, D.M.; Cretoiu, M.S.; Kielak, A.M.; Elsas, J.D. The great screen anomaly—A new frontier in product discovery through functional metagenomics. Appl. Microbiol. Biotechnol. 2012, 93, 1005–1020. [Google Scholar] [CrossRef] [PubMed]
- Najah, M.; Calbrix, R.; Mahendra-Wijaya, I.P.; Beneyton, T.; Griffiths, A.D.; Drevelle, A. Droplet-based microfluidics platform for ultra-high-throughput bioprospecting of cellulolytic microorganisms. Chem. Biol. 2014, 21, 1722–1732. [Google Scholar] [CrossRef] [PubMed]







© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Autour, A.; Ryckelynck, M. Ultrahigh-Throughput Improvement and Discovery of Enzymes Using Droplet-Based Microfluidic Screening. Micromachines 2017, 8, 128. https://doi.org/10.3390/mi8040128
Autour A, Ryckelynck M. Ultrahigh-Throughput Improvement and Discovery of Enzymes Using Droplet-Based Microfluidic Screening. Micromachines. 2017; 8(4):128. https://doi.org/10.3390/mi8040128
Chicago/Turabian StyleAutour, Alexis, and Michael Ryckelynck. 2017. "Ultrahigh-Throughput Improvement and Discovery of Enzymes Using Droplet-Based Microfluidic Screening" Micromachines 8, no. 4: 128. https://doi.org/10.3390/mi8040128
APA StyleAutour, A., & Ryckelynck, M. (2017). Ultrahigh-Throughput Improvement and Discovery of Enzymes Using Droplet-Based Microfluidic Screening. Micromachines, 8(4), 128. https://doi.org/10.3390/mi8040128