Rapid Detection and Trapping of Extracellular Vesicles by Electrokinetic Concentration for Liquid Biopsy on Chip



Abstract
:
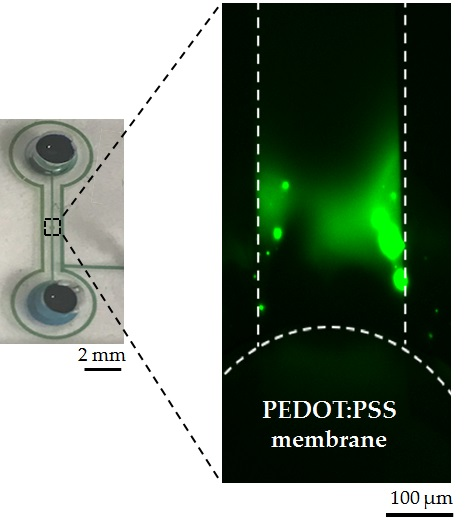{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
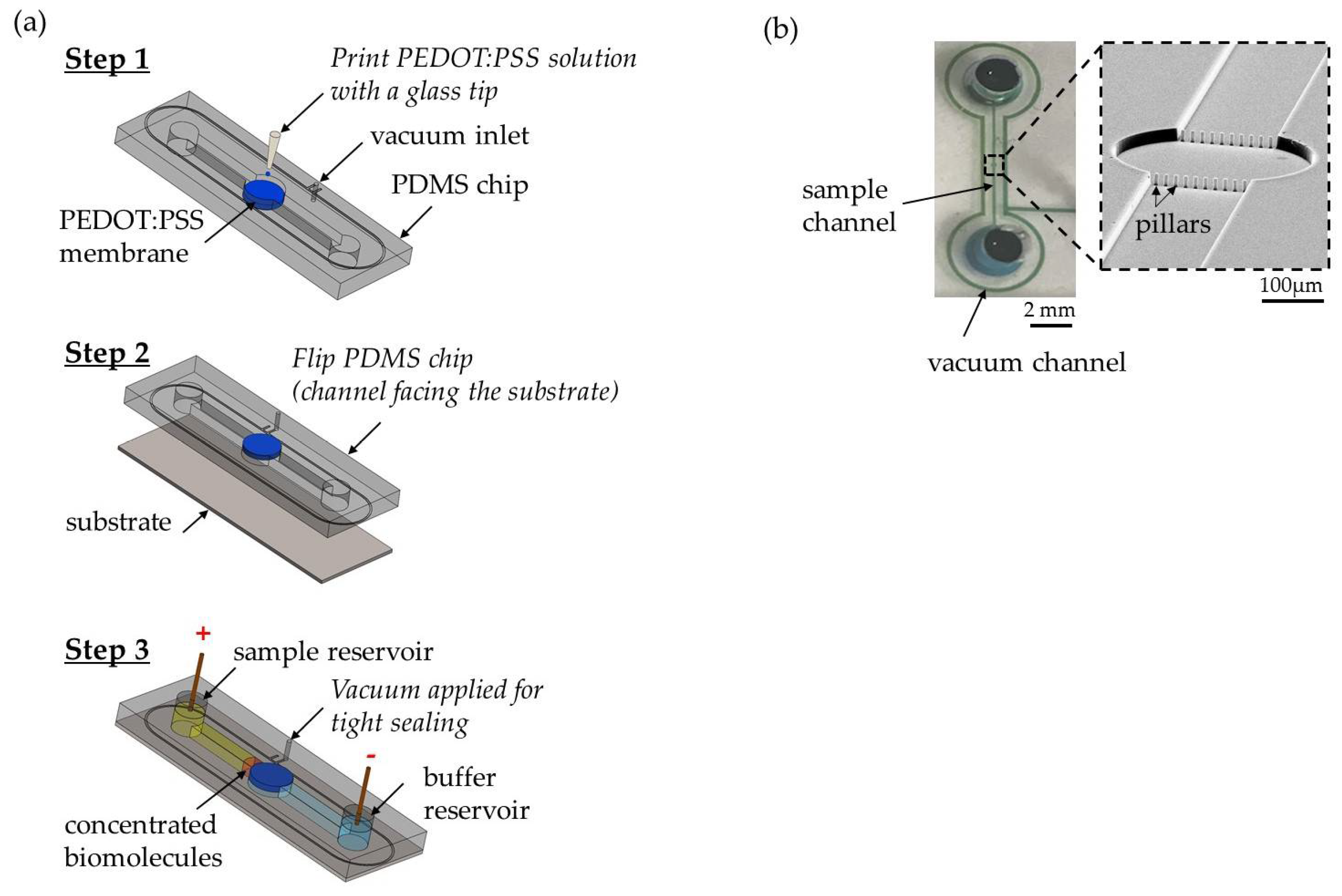
2.1. Device Fabrication and Assembly
2.2. Experiment Setup and Data Analysis
2.3. Extracellular Vesicles Extraction, Quantification, and Labeling
2.4. Characterization of Extracellular Vesicles Concentration with Fluorescence Signal Intensity without ICP
2.5. Characterization of Extracellular Vesicles with Transmission Electron Microscopy (TEM)
3. Results
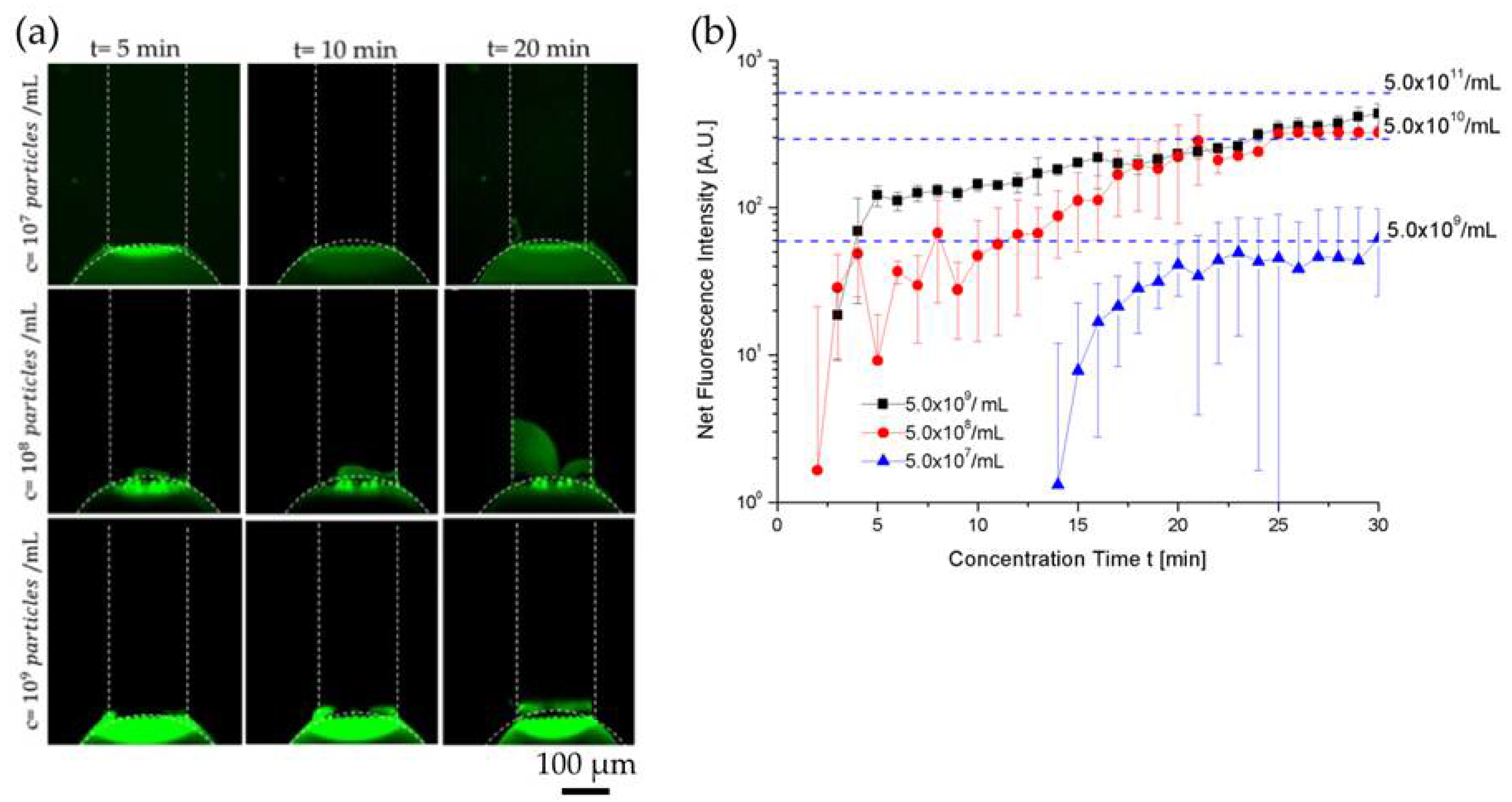
3.1. Evaluating the Enhancement in Concentration of Extracellular Vesicles
3.2. Capturing of Preconcentrated Extracellular Vesicles on an Aldehyde Substrate for Subsequent Imaging and Analysis
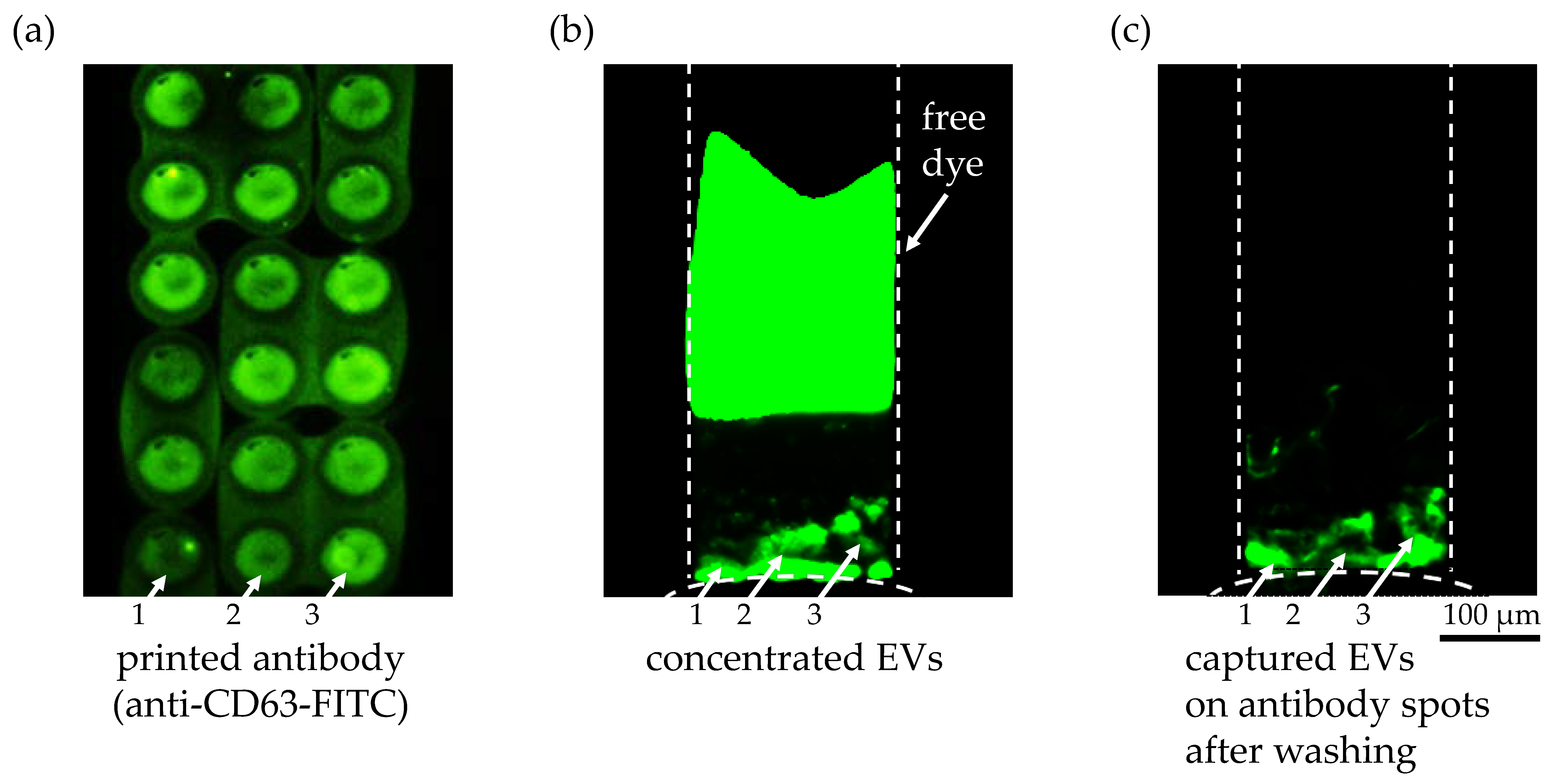
3.3. Capturing of Preconcentrated Extracellular Vesicles with Antibody Binding Assays on Super Epoxy 3 Substrate
3.4. Trapping of Preconcentrated Extracellular Vesicles with Microtraps on Glass Substrate
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Etzioni, R.; Urban, N.; Ramsey, S.; McIntosh, M.; Schwartz, S.; Reid, B.; Radich, J.; Anderson, G.; Hartwell, L. The case for early detection. Nat. Rev. Cancer 2003, 3, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Naranjo, J.C.; Wu, H.-J.; Ugaz, V.M. Microfluidics for exosome isolation and analysis: Enabling liquid biopsy for personalized medicine. Lab Chip 2017. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, A.R.; Grondin, G. Shedding of vesicular material from the cell surface of eukaryotic cells: Different cellular phenomena. BBA Rev. Biomembr. 1991, 1071, 203–219. [Google Scholar] [CrossRef]
- Andre, F.; Schartz, N.E.C.; Movassagh, M.; Flament, C.; Pautier, P.; Morice, P.; Pomel, C.; Lhomme, C.; Escudier, B.; Le Chevalier, T.; et al. Malignant effusions and immunogenic tumour-derived exosomes. Lancet 2002, 360, 295–305. [Google Scholar] [CrossRef]
- Ko, J.; Carpenter, E.; Issadore, D. Detection and isolation of circulating exosomes and microvesicles for cancer monitoring and diagnostics using micro-/nano-based devices. Analyst 2016, 141, 450–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Rai, A.J.; Joel DeCastro, G.; Zeringer, E.; Barta, T.; Magdaleno, S.; Setterquist, R.; Vlassov, A.V. An optimized procedure for exosome isolation and analysis using serum samples: Application to cancer biomarker discovery. Methods 2015, 87, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Abramowicz, A.; Widlak, P.; Pietrowska, M. Proteomic analysis of exosomal cargo: The challenge of high purity vesicle isolation. Mol. BioSyst. 2016, 12, 1407–1419. [Google Scholar] [CrossRef] [PubMed]
- Cvjetkovic, A.; Lötvall, J.; Lässer, C. The influence of rotor type and centrifugation time on the yield and purity of extracellular vesicles. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Van Deun, J.; Mestdagh, P.; Sormunen, R.; Cocquyt, V.; Vermaelen, K.; Vandesompele, J.; Bracke, M.; De Wever, O.; Hendrix, A. The impact of disparate isolation methods for extracellular vesicles on downstream RNA profiling. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momen-Heravi, F.; Balaj, L.; Alian, S.; Mantel, P.Y.; Halleck, A.E.; Trachtenberg, A.J.; Soria, C.E.; Oquin, S.; Bonebreak, C.M.; Saracoglu, E.; et al. Current methods for the isolation of extracellular vesicles. Biol. Chem. 2013, 394, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.L.; Khosroheidari, M.; Kanchi Ravi, R.; Distefano, J.K. Comparison of protein, microRNA, and mRNA yields using different methods of urinary exosome isolation for the discovery of kidney disease biomarkers. Kidney Int. 2012, 82, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 3.22.1–3.22.19. [Google Scholar] [CrossRef] [PubMed]
- Paolini, L.; Zendrini, A.; Di Noto, G.; Busatto, S.; Lottini, E.; Radeghieri, A.; Dossi, A.; Caneschi, A.; Ricotta, D.; Bergese, P. Residual matrix from different separation techniques impacts exosome biological activity. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Skog, J.; Hsu, C.-H.; Lessard, R.T.; Balaj, L.; Wurdinger, T.; Carter, B.S.; Breakefield, X.O.; Toner, M.; Irimia, D. Microfluidic isolation and transcriptome analysis of serum microvesicles. Lab Chip 2010, 10, 505–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudani, J.S.; Gossett, D.R.; Tse, H.T.K.; Lamm, R.J.; Kulkarni, R.P.; Carlo, D. Di Rapid inertial solution exchange for enrichment and flow cytometric detection of microvesicles. Biomicrofluidics 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Yang, Y.; Zeng, Y.; He, M. A microfluidic ExoSearch chip for multiplexed exosome detection towards blood-based ovarian cancer diagnosis. Lab Chip 2016, 16, 489–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reátegui, E.; Van Der Vos, K.E.; Lai, C.P.; Zeinali, M.; Atai, N.A.; Aldikacti, B.; Floyd, F.P.; Khankhel, A.; Thapar, V.; Hochberg, F.H.; et al. Engineered nanointerfaces for microfluidic isolation and molecular profiling of tumor-specific extracellular vesicles. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Davies, R.T.; Kim, J.; Jang, S.C.; Choi, E.-J.; Gho, Y.S.; Park, J. Microfluidic filtration system to isolate extracellular vesicles from blood. Lab Chip 2012, 12, 5202–5210. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Jo, W.; Heo, Y.; Kang, J.Y.; Kwak, R.; Park, J. Isolation of extracellular vesicle from blood plasma using electrophoretic migration through porous membrane. Sens. Actuators B Chem. 2016, 233, 289–297. [Google Scholar] [CrossRef]
- Marczak, S.; Richards, K.; Ramshani, Z.; Smith, E.; Senapati, S.; Hill, R.; Go, D.B.; Chang, H.C. Simultaneous isolation and preconcentration of exosomes by ion concentration polarization. Electrophoresis 2018. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Hosokawa, K.; Maeda, M. Alternating current cloud point extraction on a microchip for preconcentration of membrane-associated biomolecules. Lab Chip 2009, 9, 1168–1170. [Google Scholar] [CrossRef] [PubMed]
- Myers, G.A.; Harris, J.M. Confocal raman microscopy of pH-gradient-based 10 000-fold preconcentration of compounds within individual, optically trapped phospholipid vesicles. Anal. Chem. 2011, 83, 6098–6105. [Google Scholar] [CrossRef] [PubMed]
- Wandstrat, M.M.; Cox, J.A. Preconcentration and determination of a phospholipid at a surface modified by layer-by-layer assembly. Electroanalysis 2007, 19, 49–54. [Google Scholar] [CrossRef]
- Lee, S.J.; Rhee, H.; Jeon, T.J.; Kim, D. Preconcentration of lipid vesicles using concentration polarization in a microfluidic chip. Sens. Actuators B Chem. 2016, 229, 276–280. [Google Scholar] [CrossRef]
- Martins, D.; Levicky, R.; Song, Y.A. Enhancing the speed of morpholino-DNA biosensor by electrokinetic concentration of DNA in a microfluidic chip. Biosens. Bioelectron. 2015, 72, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Cheung, L.S.; Wei, X.; Martins, D.; Song, Y.A. Rapid detection of exosomal microRNA biomarkers by electrokinetic concentration for liquid biopsy on chip. Biomicrofluidics 2018, 12. [Google Scholar] [CrossRef]
- Lane, R.E.; Korbie, D.; Anderson, W.; Vaidyanathan, R.; Trau, M. Analysis of exosome purification methods using a model liposome system and tunable-resistive pulse sensing. Sci. Rep. 2015, 5, 7639. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheung, L.S.; Sahloul, S.; Orozaliev, A.; Song, Y.-A. Rapid Detection and Trapping of Extracellular Vesicles by Electrokinetic Concentration for Liquid Biopsy on Chip. Micromachines 2018, 9, 306. https://doi.org/10.3390/mi9060306
Cheung LS, Sahloul S, Orozaliev A, Song Y-A. Rapid Detection and Trapping of Extracellular Vesicles by Electrokinetic Concentration for Liquid Biopsy on Chip. Micromachines. 2018; 9(6):306. https://doi.org/10.3390/mi9060306
Chicago/Turabian StyleCheung, Lucia S., Sarah Sahloul, Ajymurat Orozaliev, and Yong-Ak Song. 2018. "Rapid Detection and Trapping of Extracellular Vesicles by Electrokinetic Concentration for Liquid Biopsy on Chip" Micromachines 9, no. 6: 306. https://doi.org/10.3390/mi9060306
APA StyleCheung, L. S., Sahloul, S., Orozaliev, A., & Song, Y.-A. (2018). Rapid Detection and Trapping of Extracellular Vesicles by Electrokinetic Concentration for Liquid Biopsy on Chip. Micromachines, 9(6), 306. https://doi.org/10.3390/mi9060306