Crystal Structure and Computational Study on Methyl-3-Aminothiophene-2-Carboxylate

 ,
,
Abstract
:1. Introduction
2. Experimental Details
2.1. Materials and Methods
2.2. Computational Methods
3. Results and Discussion
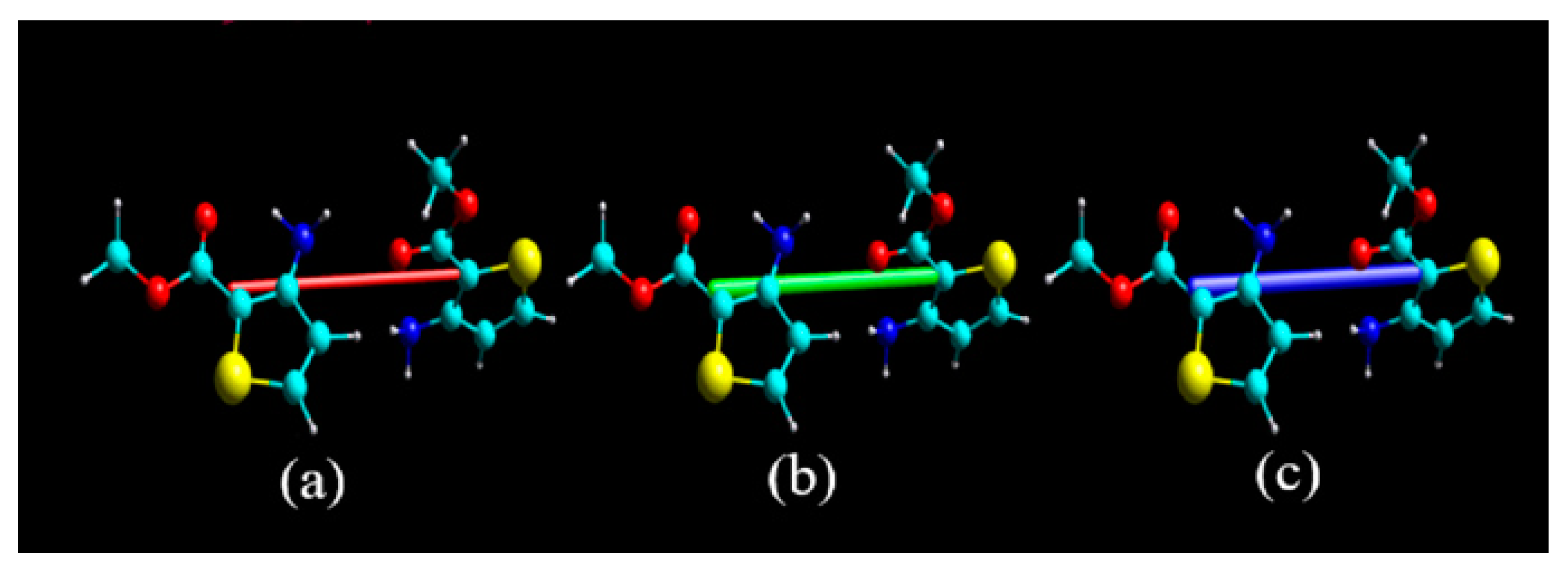
3.1. Crystal Structural Descriptions
3.2. Hirshfeld Surface Analysis and Fingerprint Plots
3.3. Reduced Density Gradient Analysis
3.4. Electrostatic Potential Surface and Frontier Molecular Orbitals Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sabri, S.S.; El-Abadla, N.S.; El-Abadelah, M.M.; Voelter, W. Synthesis and Properties of Some New 1,4-Dihydrothieno [3,2-e][1,2,4] triazepin-5-ones. Z. Nat. 2006, 61, 65–68. [Google Scholar]
- Russell, R.K.; Press, J.B.; Rampulla, R.A.; McNally, J.J.; Falotico, R.; Keiser, J.A.; Brigh, D.A.; Tobia, A. Thiophene systems. 9. Thienopyrimidinedione derivatives as potential antihypertensive agents. J. Med. Chem. 1988, 31, 1786–1793. [Google Scholar] [CrossRef] [PubMed]
- Snégaroff, K.; Lassagne, F.; Bentabed-Ababsa, G.; Nassar, E.; Ely, S.C.S.; Hesse, S.; Perspicace, E.; Derdour, A.; Mongin, F. Direct metallation of thienopyrimidines using a mixed lithium–cadmium base and antitumor activity of functionalized derivatives. Org. Biomol. Chem. 2009, 7, 4782–4788. [Google Scholar] [CrossRef] [Green Version]
- Brzozowski, Z.; Sławiński, J.; Sączewski, F.; Sanchez, T.; Neamati, N. Synthesis, anti-HIV-1 integrase, and cytotoxic activities of 4-chloro-N-(4-oxopyrimidin-2-yl)-2-mercaptobenzenesulfonamide derivatives. Eur. J. Med. Chem. 2008, 43, 1188–1198. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Liu, X.; Yan, R.; Li, J.; Pannecouque, C.; Witvrouw, M.; de Clercq, E. Synthesis and anti-HIV evaluation of novel 1,3-disubstituted thieno [3,2-c][1,2,6] thiadiazin-4(3H)-one 2,2-dioxides (TTDDs). Bioorganic Med. Chem. 2008, 16, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Lazerwith, S.E.; Lew, W.; Zhang, J.; Morganelli, P.; Liu, Q.; Canales, E.; Clarke, M.O.; Doerffler, E.; Byun, D.; Mertzman, M.; et al. Discovery of GS-9669, a thumb site II non-nucleoside inhibitor of NS5B for the treatment of genotype 1 chronic hepatitis C infection. J. Med. Chem. 2013, 57, 1893–1901. [Google Scholar] [CrossRef] [PubMed]
- Wiley, M.R.; Weir, L.C.; Briggs, S.; Bryan, N.A.; Buben, J.; Campbell, C.; Chirgadze, N.Y.; Conrad, R.C.; Craft, T.J.; Ficorilli, J.V.; et al. Structure-based design of potent, amidine-derived inhibitors of factor Xa: Evaluation of selectivity, anticoagulant activity, and antithrombotic activity. J. Med. Chem. 2000, 43, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.W.; Guo, C.; Piraino, J.; Westwick, J.K.; Zhang, C.; Lamerdin, J.; Dagostino, E.; Knighton, D.; Loi, C.M.; Zager, M.; et al. Small-molecule p21-activated kinase inhibitor PF-3758309 is a potent inhibitor of oncogenic signaling and tumor growth. Proc. Natl. Acad. Sci. USA 2010, 107, 9446–9451. [Google Scholar] [CrossRef] [Green Version]
- Tian, Q.; Cheng, Z.; Yajima, H.M.; Savage, S.J.; Green, K.L.; Humphries, T.; Reynolds, M.E.; Babu, S.; Gosselin, F.; Askin, D.; et al. A practical synthesis of a PI3K inhibitor under noncryogenic conditions via functionalization of a lithium triarylmagnesiate intermediate. Org. Process Res. Dev. 2013, 17, 97–107. [Google Scholar] [CrossRef]
- Rizwan, K.; Zubair, M.; Rasool, N.; Ali, S.; Zahoor, A.F.; Rana, U.A.; Khan, S.U.D.; Shahid, M.; Jaafar, H.Z. Regioselective synthesis of 2-(bromomethyl)-5-aryl-thiophene derivatives via palladium (0) catalyzed suzuki cross-coupling reactions: As antithrombotic and haemolytically active molecules. Chem. Cent. J. 2014, 8, 74. [Google Scholar] [CrossRef] [Green Version]
- Labat, Y. Thioglycolic Acid. In Kirk-Othmer Encyclopedia of Chemical Technology; Wiley: Hoboken, NJ, USA, 2000. [Google Scholar]
- Baumgartner, T. π-Conjugated Heterocyclic fused Bithiophene Materials. J. Inorg. Organomet. Polym. Mater. 2005, 15, 389–409. [Google Scholar] [CrossRef]
- Fuller, L.S. Thiophene and thiophene derivatives. In Kirk-Othmer Encyclopedia of Chemical Technology; Wiley: Hoboken, NJ, USA, 2000. [Google Scholar]
- Huddleston, P.R.; Barker, J.M. A Convenient Synthesis of 2-Substituted 3-Hydroxy-and 3-Amino-Thiophens from Derivatives of 2-Chloroacrylic Acid. Synth. Commun. 1979, 9, 731–734. [Google Scholar] [CrossRef]
- Rigaku, O.D. CrysAlis PRO; Rigaku Oxford Diffraction: Yarnton, UK, 2015. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, 71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D. 01; Gaussian. Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer17.5; The University of Western Australia: Perth, Australia, 2017. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Alizadeh, A.; Vahabi, A.H.; Bazgir, A.; Khavasi, H.R.; Zhu, Z.; Zhu, L.G. Determinative role of ring size and substituents in highly selective synthesis of functionalized bicyclic guanidine and tetra substituted thiophene derivatives based on salt adducts afforded by cyclic thioureas and ketene dithioacetal. Tetrahedron 2016, 72, 1342–1350. [Google Scholar] [CrossRef]
- Sahu, S.N.; Gupta, M.K.; Singh, S.S.; Yadav, P.; Panwar, R.; Kumar, A.; Ram, V.J.; Kumar, B.; Pratap, R. One pot synthesis of tetrasubstituted thiophenes: [3 + 2] annulation strategy. RSC Adv. 2015, 5, 36979–36986. [Google Scholar] [CrossRef]
- Alizadeh, A.; Moafi, L.; Ghanbaripour, R.; Abadi, M.H.; Zhu, Z.; Kubicki, M. A new route for the synthesis of 1,3,4-trisubstituted pyrazolo[4,3-c]quinolines via a multicomponent reaction. Tetrahedron 2015, 71, 3495–3499. [Google Scholar] [CrossRef]
- Singh, H. A DFT approach for theoretical and experimental study of structure, electronic, Hirshfeld surface and spectroscopic properties of 12-(4-bromophenyl)-2-(prop-2-ynyloxy)-9,10-dihydro-8H-benzo [a]xanthen-11(12H)-on single crystal. Chem. Phys. 2019, 524, 1–13. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Fabbiani, F.P.A.; Spackman, M.A. Comparison of polymorphic molecular crystal structures through Hirshfeld surface analysis. Cryst. Growth Des. 2007, 7, 755–769. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr. Sect. B Struct. Sci. 2004, 60, 627–668. [Google Scholar] [CrossRef] [PubMed]
- Hemissi, H.; Nasri, A.; Rzaigui, M. Chiral self-assembly of a novel (CuII/ReVII)-heterobimetallic l-Arginine complex: Crystal structure, Hirshfeld surface analysis, spectroscopic properties and biochemical studies. J. Mol. Struct. 2019, 1186, 307–316. [Google Scholar] [CrossRef]
- Tan, X.J.; Zhang, L.Y.; Sun, Y.K.; Zhou, X.M. Synthesis, structure and antiproliferative and optical activities of two new biphenyl-derived Schiff bases. Acta Cryst. 2019, 75, 97–106. [Google Scholar] [CrossRef]
- Gumus, I.; Solmaz, U.; Binzet, G.; Keskin, E.; Arslan, B.; Arslan, H. Hirshfeld surface analyses and crystal structures of supramolecular self-assembly thiourea derivatives directed by non-covalent interactions. J. Mol. Struct. 2018, 1157, 78–88. [Google Scholar] [CrossRef]
- Edwards, A.J.; Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Intermolecular interactions in molecular crystals: What’s in a name? Faraday Discuss. 2017, 203, 93–112. [Google Scholar] [CrossRef] [Green Version]
- Geetha, D.V.; Al-Ostoot, F.H.; Mohammed, Y.H.E.; Sridhar, M.A.; Khanum, S.A.; Lokanath, N.K. Synthesis, Elucidation, Hirshfeld surface analysis, and DFT calculations of 4-chloro-N-[2-(2-1H-indol-3-yl-acetylamino)-phenyl]-benzamide. J. Mol. Struct. 2019, 1178, 384–393. [Google Scholar] [CrossRef]
- Tao, Y.; Han, L.; Li, X.; Han, Y.; Liu, Z. Molecular structure, spectroscopy (FT-IR, FT-Raman), thermodynamic parameters, molecular electrostatic potential and HOMO-LUMO analysis of 2, 6-dichlorobenzamide. J. Mol. Struct. 2016, 1108, 307–314. [Google Scholar] [CrossRef]
- Lu, T.; Manzetti, S. Wavefunction and reactivity study of benzo [a] pyrene diol epoxide and its enantiomeric forms. Struct. Chem. 2014, 25, 1521–1533. [Google Scholar] [CrossRef]
- Tao, Y.; Han, L.; Han, Y.; Liu, Z. A combined experimental and theoretical analysis on molecular structure and vibrational spectra of 2, 4-dihydroxybenzoic acid. Spectrochim. Acta 2015, 137, 1078–1085. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Li, X.; Han, L.; Zhang, W.; Liu, Z. Spectroscopy (FT-IR, FT-Raman), hydrogen bonding, electrostatic potential and HOMO-LUMO analysis of tioxolone based on DFT calculations. J. Mol. Struct. 2016, 1121, 188–195. [Google Scholar] [CrossRef]
- Qi, Y.; Tang, Z.; Zhan, H.; Wang, Y.; Zhao, Y.; Fei, X.; Tian, J.; Yu, L.; Liu, J. A new interpretation of the ESIPT mechanism of 2-(benzimidazol-2-yl)-3-hydroxychromone derivatives. Spectrochim. Acta 2020, 224, 117359. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Lu, M.; Wang, Y.; Tang, Z.; Gao, Z.; Tian, J.; Fei, X.; Li, Y.; Liu, J. A theoretical study of the ESIPT mechanism of 3-hydroxyflavone derivatives: Solvation effect and the importance of TICT for its dual fluorescence properties. Org. Chem. Front. 2019, 6, 3136–3143. [Google Scholar] [CrossRef]
- Yin, X.; Lai, X.Y.; Wang, X.; Liu, Y.T. Theoretical insight into the photophysical properties of long-lifetime Ir (iii) and Rh (iii) complexes for two-photon photodynamic therapy. Phys. Chem. Chem. Phys. 2019, 21, 8394–8401. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CCDC Number | 1946321 |
|---|---|
| Empirical formula | C6H7NO2S |
| Formula weight | 157.19 |
| Temperature/K | 100 |
| Crystal system | monoclinic |
| Crystal size/mm3 | 0.26 × 0.25 × 0.22 |
| Space group | P21/c |
| a, b, and c/Å | 14.7315(2), 6.1118(1), 24.2439(3) |
| Β/° | 95.984(1) |
| Volume/Å3 | 2170.93(5) |
| Z | 12 |
| ρcalc g/cm3 | 1.443 |
| μ/mm−1 | 3.480 |
| F(000) | 984.0 |
| 2Θ range for data collection/° | 6.032 to 151.934 |
| Index ranges | ‒18 ≤ h ≤ 18, ‒7 ≤ k ≤ 4, ‒30 ≤ l ≤ 28 |
| Reflections collected | 20121 |
| Independent reflections | 4440 [Rint = 0.0457, Rsigma = 0.0352] |
| Data/restraints/parameters | 4440/0/274 |
| Goodness-of-fit on F2 | 1.058 |
| Final R indexes [I > = 2σ (I)] | R1 = 0.0384, wR2 = 0.1040 |
| Final R indexes [all data] | R1 = 0.0423, wR2 = 0.1094 |
| Largest diff. peak/hole/e Å−3 | 0.46/−0.34 |
| Donor–H⋯Acceptor | Distance of D–H/Å | Distance of H⋯A/Å | Distance of D⋯A/Å | Angle of D–H⋯A/˚ |
|---|---|---|---|---|
| N1H1A···O2 | 0.85 | 2.24 | 2.836(2) | 127 |
| N1–H1B···O4 i | 0.84 | 2.06 | 2.8827(19) | 169 |
| N2–H2B···O4 | 0.80 | 2.31 | 2.8644(19) | 127 |
| N3–H3D···O2 ii | 0.82 | 2.11 | 2.895(2) | 159 |
| N3–H3E···O5 | 0.86 | 2.25 | 2.830(2) | 125 |
| N2–H2B···N3 iii | 0.80 | 2.49 | 3.195(2) | 147 |
| C9–H9B···S1 | 0.98 | 2.86 | 3.5794(18) | 131 |
| C3–H3C···Cg1 iv | 0.98 | 2.79 | 3.655(2) | 148 |
| C5–H5···Cg1 ii | 0.95 | 2.92 | 3.569(2) | 126 |
| C9–H9C···Cg2 iv | 0.98 | 2.67 | 3.4942(19) | 142 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tao, Y.; Han, L.; Sun, A.; Sun, K.; Zhang, Q.; Liu, W.; Du, J.; Liu, Z. Crystal Structure and Computational Study on Methyl-3-Aminothiophene-2-Carboxylate. Crystals 2020, 10, 19. https://doi.org/10.3390/cryst10010019
Tao Y, Han L, Sun A, Sun K, Zhang Q, Liu W, Du J, Liu Z. Crystal Structure and Computational Study on Methyl-3-Aminothiophene-2-Carboxylate. Crystals. 2020; 10(1):19. https://doi.org/10.3390/cryst10010019
Chicago/Turabian StyleTao, Yaping, Ligang Han, Andong Sun, Kexi Sun, Qian Zhang, Wanqiang Liu, Jianbin Du, and Zhaojun Liu. 2020. "Crystal Structure and Computational Study on Methyl-3-Aminothiophene-2-Carboxylate" Crystals 10, no. 1: 19. https://doi.org/10.3390/cryst10010019