Sildenafil–Resorcinol Cocrystal: XRPD Structure and DFT Calculations





Abstract
1. Introduction
2. Materials, Experimental, and Theoretical Methods
2.1. Synthesis
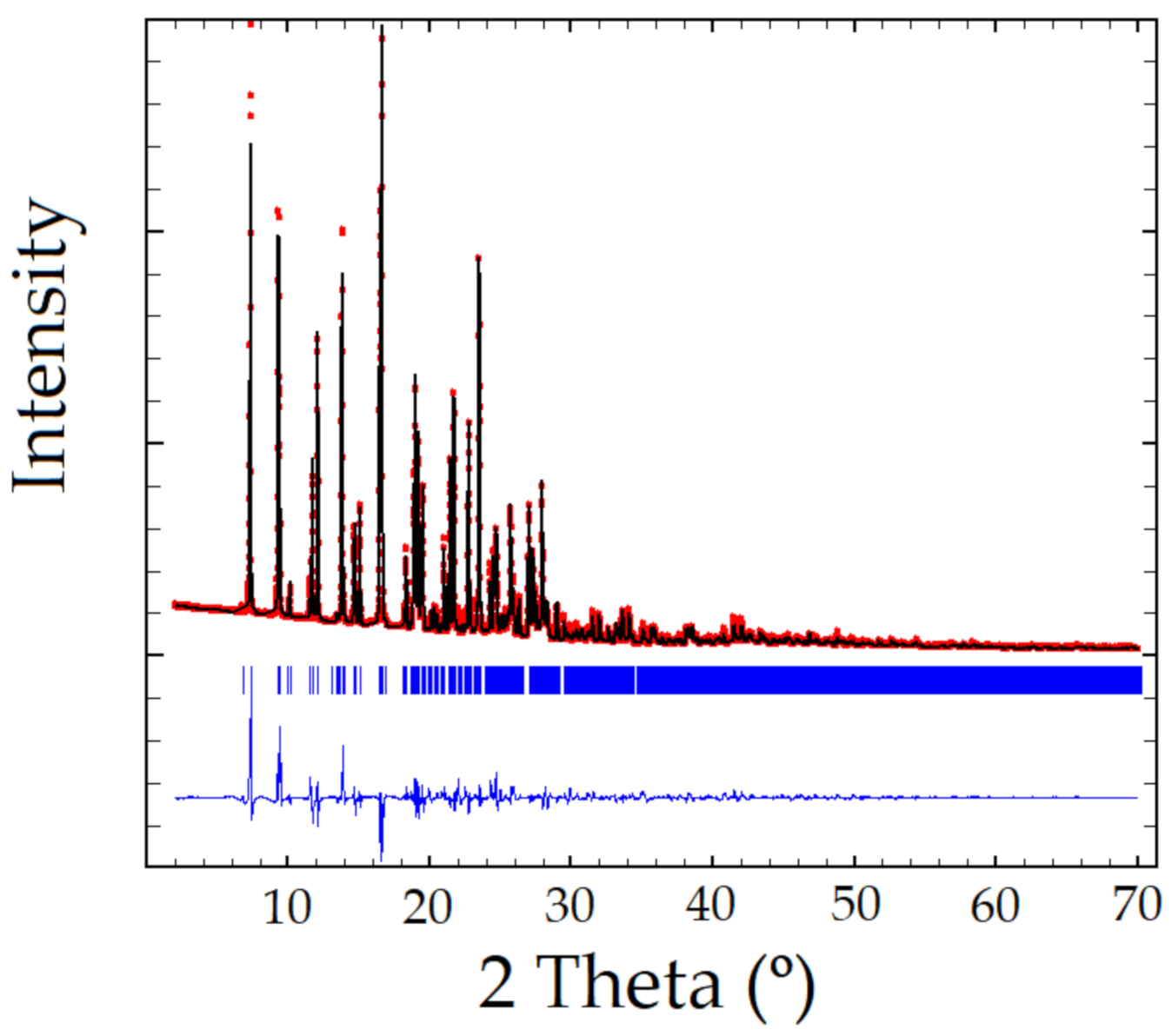
2.2. XRPD Analysis
2.3. Theoretical Methods
3. Results and Discussion
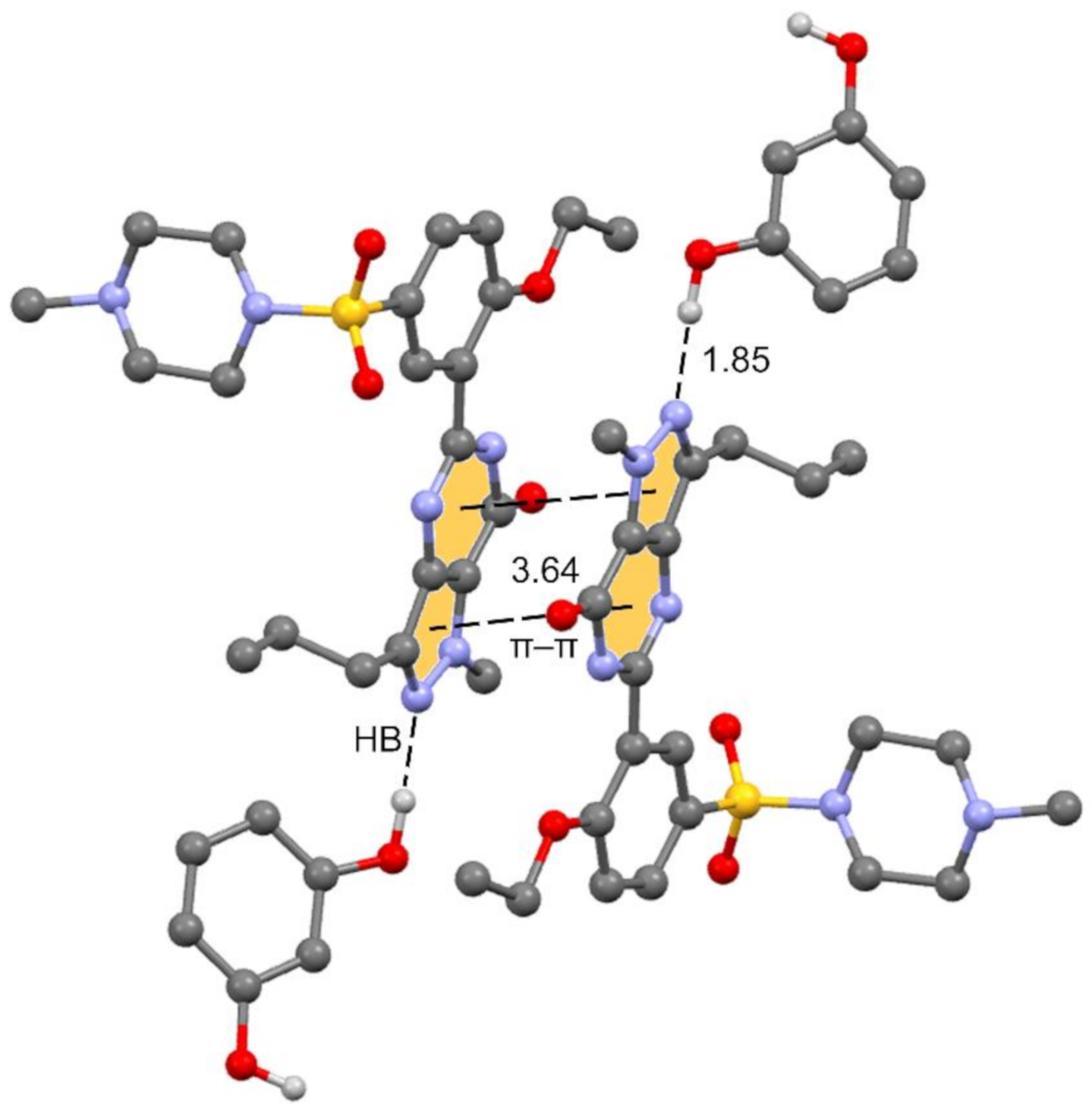
3.1. Description of the Cocrystal
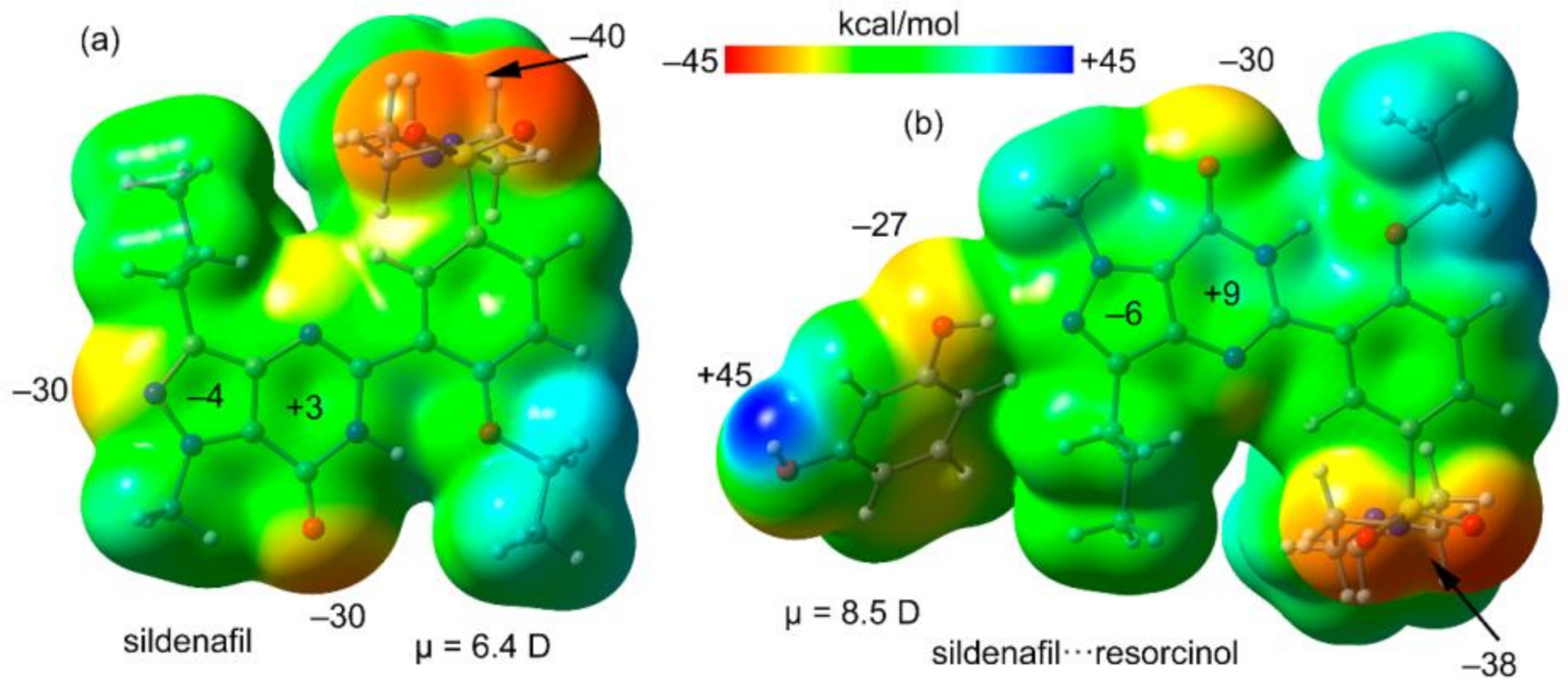
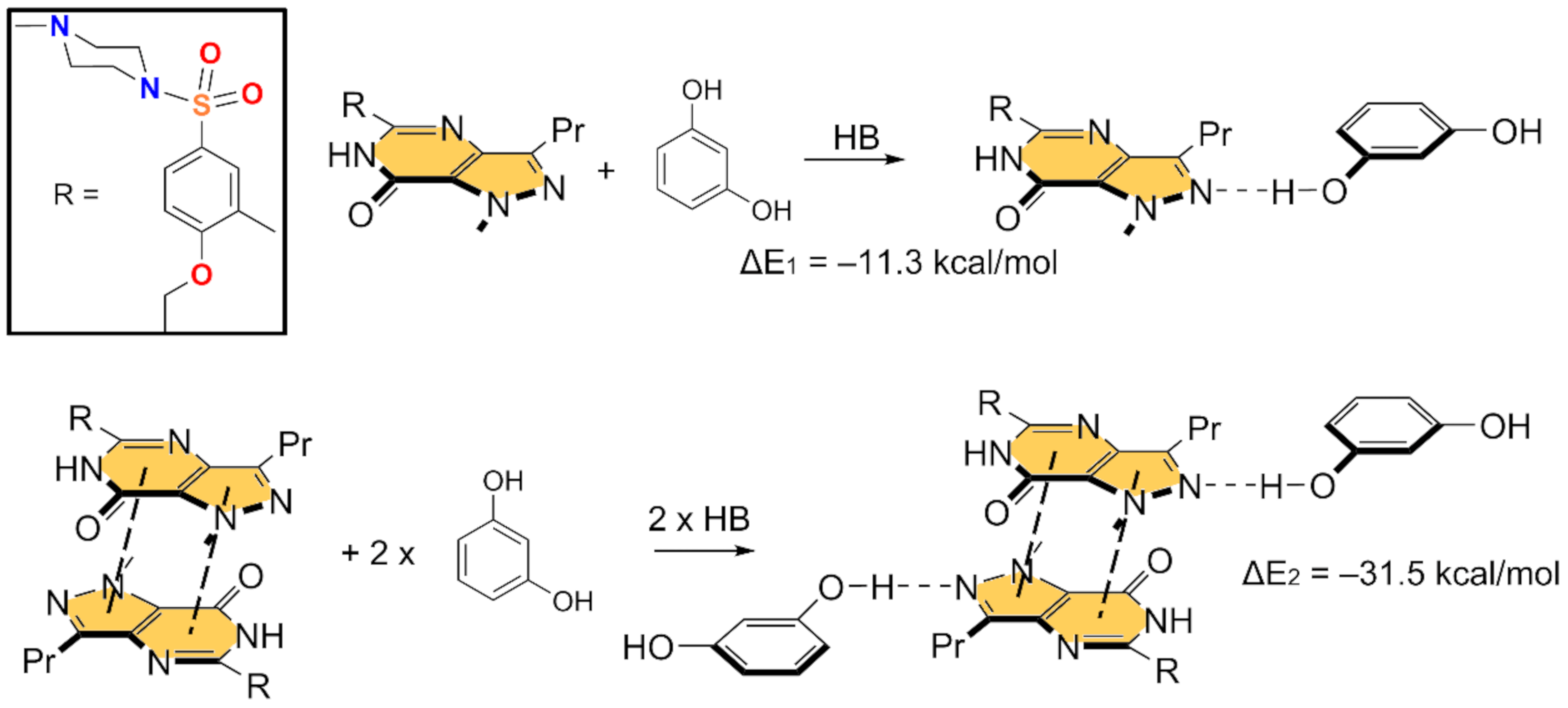
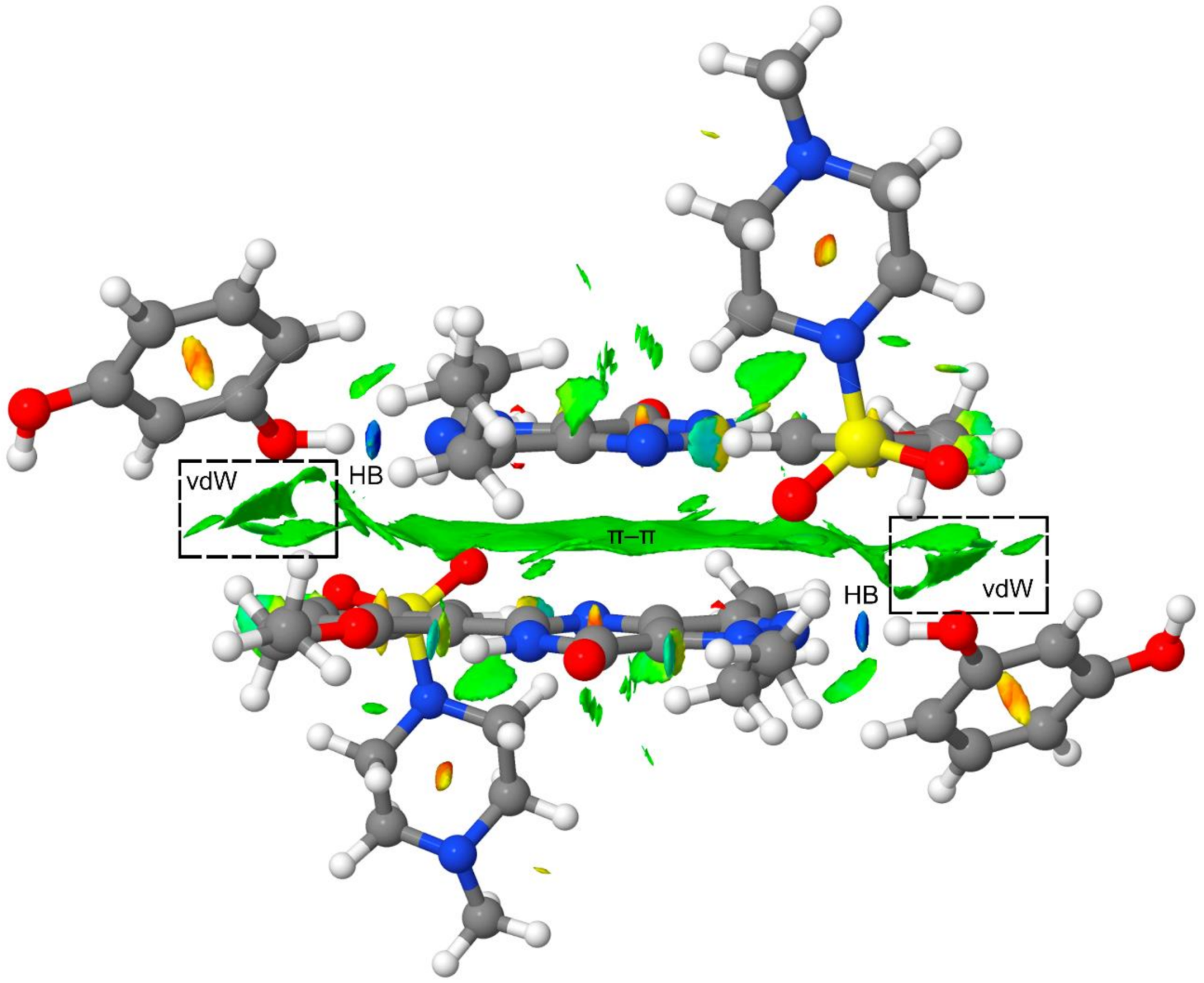
3.2. Theoretical Study
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Boolell, M.; Allen, M.J.; Ballard, S.A.; Gepi-Attee, S.; Muirhead, G.J.; Naylor, A.M.; Osterloh, I.O.; Gingell, C. Sildenafil: An orally active type 5 cyclic GMP-specific phosphodiesterase inhibitor for the treatment of penile erectile dysfunction. Int. J. Impot. Res. 1996, 8, 47–52. [Google Scholar]
- Ballard, S.A.; Gingell, C.J.; Tang, K.; Turner, L.A.; Price, M.E.; Naylor, A.M. Effects of Sildenafil on the relaxation of human corpus cavernosum tissue in vitro and on the activities of cyclic nucleotide phosphodiesterase isozymes. J. Urol. 1998, 159, 2164–2171. [Google Scholar] [CrossRef]
- Yathirajan, H.S.; Nagaraj, B.; Nagaraja, P.; Bolte, M. Sildenafil citrate monohydrate. Acta Crystallogr. Sect. E: Struct. Rep. Online 2005, 61, 489–491. [Google Scholar] [CrossRef]
- Stepanovs, D.; Mishnev, A. Molecular and crystal structure of Sildenafil base. Z. Naturforsch. B: J. Chem. Sci. 2012, 67, 491–494. [Google Scholar] [CrossRef]
- Banerjee, R.; Bhatt, P.M.; Desiraju, G.R. Solvates of Sildenafil Saccharinate. A New Host Material. Cryst. Growth Des. 2006, 6, 1468–1478. [Google Scholar] [CrossRef]
- Sanphui, P.; Tothadi, S.; Ganguly, S.; Desiraju, G.R. Salt and Cocrystals of Sildenafil with Dicarboxylic Acids: Solubility and Pharmacokinetic Advantage of the Glutarate Salt. Mol. Pharm. 2013, 10, 4687–4697. [Google Scholar] [CrossRef] [PubMed]
- Žegarac, M.; Lekšic, E.; Šket, P.; Plavec, J.; Bogdanovic, M.D.; Bucar, D.-K.; Dumic, M.; Meštrovic, E.A. Sildenafil cocrystal based on acetylsalicylic acid exhibits an enhanced intrinsic dissolution rate. CrystEngComm 2014, 16, 32–35. [Google Scholar] [CrossRef]
- Stepanovs, D.; Jureb, M.; Mishnev, A. Preparation and crystal structure of Sildenafil salicylate. Mendeleev Commun. 2015, 25, 49–50. [Google Scholar] [CrossRef]
- Barbas, R.; Font-Bardia, M.; Paradkar, A.; Hunter, C.A.; Prohens, R. Combined Virtual/Experimental Multicomponent Solid Forms Screening of Sildenafil: New Salts, Cocrystals, and Hybrid Salt−Cocrystals. Cryst. Growth Des. 2018, 18, 7618–7627. [Google Scholar] [CrossRef]
- Barbas, R.; Font-Bardia, M.; Prohens, R. Polymorphism of Sildenafil: A New Metastable Desolvate. Cryst. Growth Des. 2018, 18, 3740–3746. [Google Scholar] [CrossRef]
- Barbas, R.; Prohens, R.; Font-Bardía, M.; Bauzá, A.; Frontera, A. Hydrogen bonding versus π-interactions: Their key competition in Sildenafil solvates. CrystEngComm 2018, 20, 4526–4530. [Google Scholar] [CrossRef]
- Wang, C.; Perumalla, S.R.; Sun, C.C. Anion Exchange Reaction for Preparing Acesulfame Solid Forms. Cryst. Growth Des. 2018, 18, 4215–4219. [Google Scholar] [CrossRef]
- Byrn, S.R.; Zografi, G.; Chen, X. Solid-State Properties of Pharmaceutical Materials, Chapter 3: Solvates and Hydrates; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2017. [Google Scholar]
- Rodríguez-Spong, B.; Price, C.P.; Jayasankar, A.; Matzger, A.J.; Rodríguez-Hornedo, N. General principles of pharmaceutical solid polymorphism: A supramolecular perspective. Adv. Drug Delivery Rev. 2004, 56, 241–274. [Google Scholar] [CrossRef] [PubMed]
- Griesser, U.J. Polymorphism in the Pharmaceutical Industry; Hilfiker, R., von Raumer, M., Eds.; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2006. [Google Scholar]
- Byrn, S.R.; Pfeiffer, R.R.; Stowell, J.G. Solid-State Chemistry of Drugs; SSCI Inc.: West Lafayette, IN, USA, 1999. [Google Scholar]
- Morissette, S.L.; Almarsson, O.; Peterson, M.L.; Remenar, J.F.; Read, M.J.; Lemmo, A.V.; Ellis, S.; Cima, M.J.; Gardner, C.R. High-throughput crystallization: Polymorphs, salts, co-crystals and solvates of pharmaceutical solids. Adv. Drug Delivery Rev. 2004, 56, 275–300. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-Garcia, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Boultif, A.; Louër, D. Indexing of powder diffraction patterns for low-symmetry lattices by the successive dichotomy method. J. Appl. Crystallogr. 1991, 24, 987–993. [Google Scholar] [CrossRef]
- Vallcorba, O.; Rius, J.; Frontera, C.; Miravitlles, C. TALP: A multisolution direct-space strategy for solving molecular crystals from powder diffraction data based on restrained least squares. J. Appl. Crystallogr. 2012, 45, 1270–1277. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian 16; Revision, B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bar, M.; Haser, M.; Horn, H.; Kolmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154118. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. J. Mol. Phys. 1970, 19, 553–566. [Google Scholar]
- Todd, A.; Keith, T.K. AIMAll; Version 13.05.06; Gristmill Software: Overland Park, KS, USA, 2013. [Google Scholar]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Efimenko, Z.M.; Eliseeva, A.A.; Ivanov, D.M.; Galmes, B.; Frontera, A.; Bokach, N.A.; Kukushkin, V.Y. Bifurcated μ2-I⋯(N,O) Halogen Bonding: The Case of (Nitrosoguanidinate)NiII Cocrystals with Iodine(I)-Based σ-Hole Donors. Cryst. Growth Des. 2020. [Google Scholar] [CrossRef]
- Zelenkov, L.E.; Ivanov, D.M.; Sadykov, E.K.; Bokach, N.A.; Galmes, B.; Frontera, A.; Kukushkin, V.Y. Semicoordination Bond Breaking and Halogen Bond Making Change the Supramolecular Architecture of Metal-Containing Aggregates. Cryst. Growth Des. 2020, 20, 6956–6965. [Google Scholar] [CrossRef]
- Soldatova, N.S.; Postnikov, P.S.; Suslonov, V.V.; Kissler, T.Y.; Ivanov, D.M.; Yusubov, M.S.; Galmes, B.; Frontera, A.; Kukushkin, V.Y. Diaryliodonium as a double σ-hole donor: The dichotomy of thiocyanate halogen bonding provides divergent solid state arylation by diaryliodonium cations. Org. Chem. Front. 2020, 7, 2230–2242. [Google Scholar] [CrossRef]
- Katlenok, E.A.; Haukka, M.; Levin, O.V.; Frontera, A.; Kukushkin, V.Y. Supramolecular Assembly of Metal Complexes by (Aryl)I⋯dz2 [PtII] Halogen Bonds. Chem. Eur. J. 2020, 26, 7692–7701. [Google Scholar] [CrossRef]
- Rozhkov, A.V.; Eliseeva, A.A.; Baykov, S.V.; Galmes, B.; Frontera, A.; Kukushkin, V.Y. One-Pot Route to X-perfluoroarenes (X = Br, I) Based on FeIII-Assisted C–F Functionalization and Utilization of These Arenes as Building Blocks for Crystal Engineering Involving Halogen Bonding. Cryst. Growth Des. 2020, 20, 5908–5921. [Google Scholar] [CrossRef]
- Rozhkov, A.V.; Ananyev, I.V.; Gomila, R.M.; Frontera, A.; Kukushkin, V.Y. π-Hole⋯dz2[PtII] Interactions with Electron-Deficient Arenes Enhance the Phosphorescence of PtII-Based Luminophores. Inorg. Chem. 2020, 59, 9308–9314. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Structure Data | |
|---|---|
| Empirical formula | C34H42N6O8S |
| Formula Weight | 694.80 |
| Temperature (K) | 298(2) |
| Wavelength (Å) | 1.5406 |
| Crystal system | Monoclinic |
| space group | P 21/a |
| a, b, c (Å) | 14.27040(15) 26.0868(3) 9.99412(12) |
| α, β, γ (°) | 90 108.3991(7) 90 |
| Volume (Å3) | 3530.31(7) |
| Z, Density (calc.) (mg/m3) | 4, 1.307 |
| θ range for data collection (°) | 2.0 to 70 step 0.013 (2θ) |
| Refinement method | Rietveld |
| Data/restraints/parameters | 3477/52/77 |
| Final R indices [I > 2σ(I)] | Rwp = 8.61 Chi2 = 107 (Rexp = 0.834) |
| CCDC | 2044269 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbas, R.; Kumar, V.; Vallcorba, O.; Prohens, R.; Frontera, A. Sildenafil–Resorcinol Cocrystal: XRPD Structure and DFT Calculations. Crystals 2020, 10, 1126. https://doi.org/10.3390/cryst10121126
Barbas R, Kumar V, Vallcorba O, Prohens R, Frontera A. Sildenafil–Resorcinol Cocrystal: XRPD Structure and DFT Calculations. Crystals. 2020; 10(12):1126. https://doi.org/10.3390/cryst10121126
Chicago/Turabian StyleBarbas, Rafael, Vineet Kumar, Oriol Vallcorba, Rafel Prohens, and Antonio Frontera. 2020. "Sildenafil–Resorcinol Cocrystal: XRPD Structure and DFT Calculations" Crystals 10, no. 12: 1126. https://doi.org/10.3390/cryst10121126
APA StyleBarbas, R., Kumar, V., Vallcorba, O., Prohens, R., & Frontera, A. (2020). Sildenafil–Resorcinol Cocrystal: XRPD Structure and DFT Calculations. Crystals, 10(12), 1126. https://doi.org/10.3390/cryst10121126