
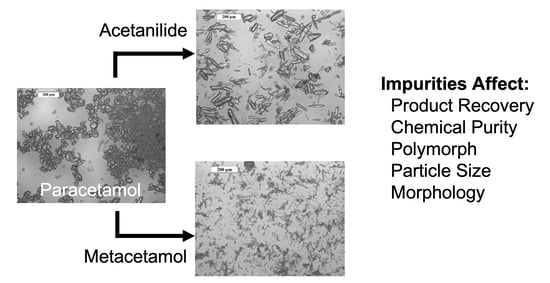
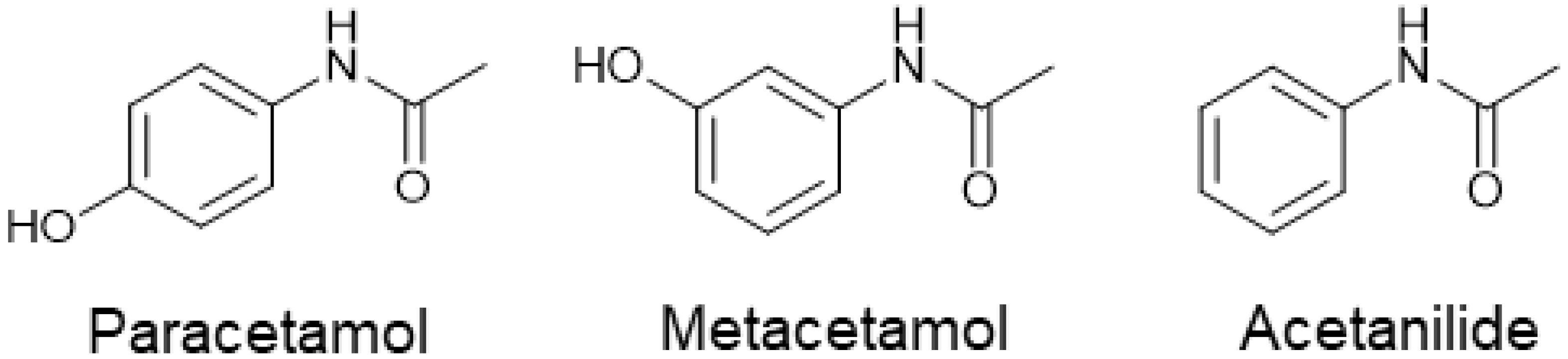
Impact of Impurities on Crystallization and Product Quality: A Case Study with Paracetamol


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
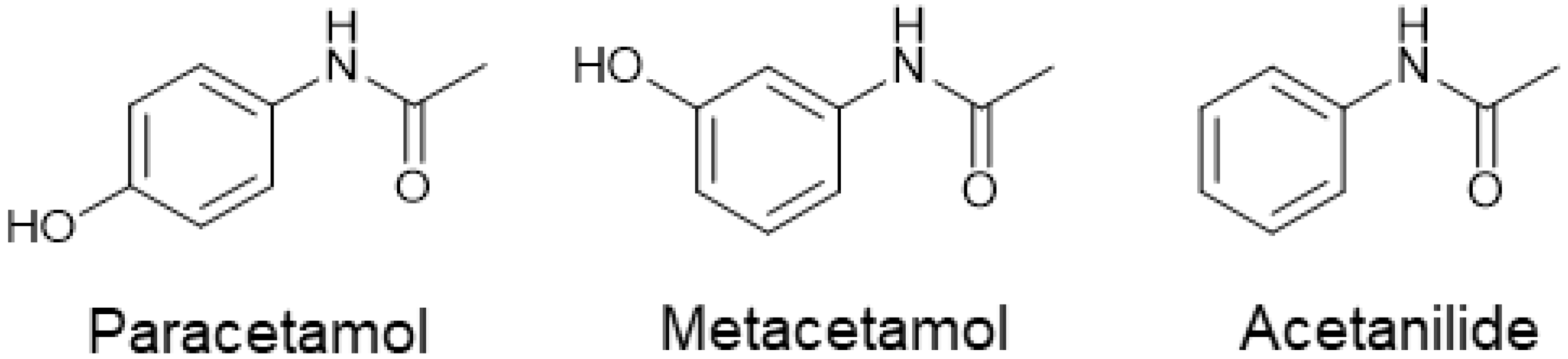
1. Introduction
2. Materials and Methods
2.1. Crystallization and Isolation
2.2. Product Analysis
2.3. List of Terms
| A | Aspect ratio |
| C | Solution concentration (mg g−1) |
| Cs | Saturation concentration at given temperature (mg g−1) |
| ΔH° | Enthalpy (J g−1) |
| I | Relative impurity spike of impurity in respect to paracetamol (%) |
| J | Nucleation rate (m−3 s−1) |
| l | Particle length (μm) |
| M | Total number nucleation experiments |
| M+ | Number of successful nucleation experiments at a particular time |
| Moles of impurity in feed (mol) | |
| Moles of API in feed (mol) | |
| Moles of impurity in crystallized solid (mol) | |
| Moles API in crystallized solid (mol) | |
| Moles of impurity in mother liquor (mol) | |
| Moles of API in crystallized solid (mol) | |
| Moles of impurity in solid after reslurry (mol) | |
| Moles of API in solid after reslurry (mol) | |
| P | Probability of nucleation |
| Yr | Product recovery (%) |
| R | Universal gas constant (J K−1 mol−1) |
| S | Supersaturation ratio |
| ΔS° | Temperature independent entropy (J K−1) |
| T | Temperature of solution saturation (°C) |
| Tm | Melting temperature (°C) |
| t | True induction time (s) |
| tind | Observed induction time (s) |
| tg | Time between nucleation occurring and observed (s) |
| V | Volume (cm3) |
| w | Particle width (μm) |
| Xa | Mole fraction of solute (-) |
| Xi,F | Impurity molar concentration in the feed (%) |
| Xi,p | Concentration of impurity in the crystallized solid (mol%) |
| Xi,s | Concentration of impurity in solid after reslurry (mol%) |
| Xp | Product purity (mol%) |
| Xp,s | Crystal purity after the reslurrying process (%) |
3. Results
3.1. Crystallization Conditions
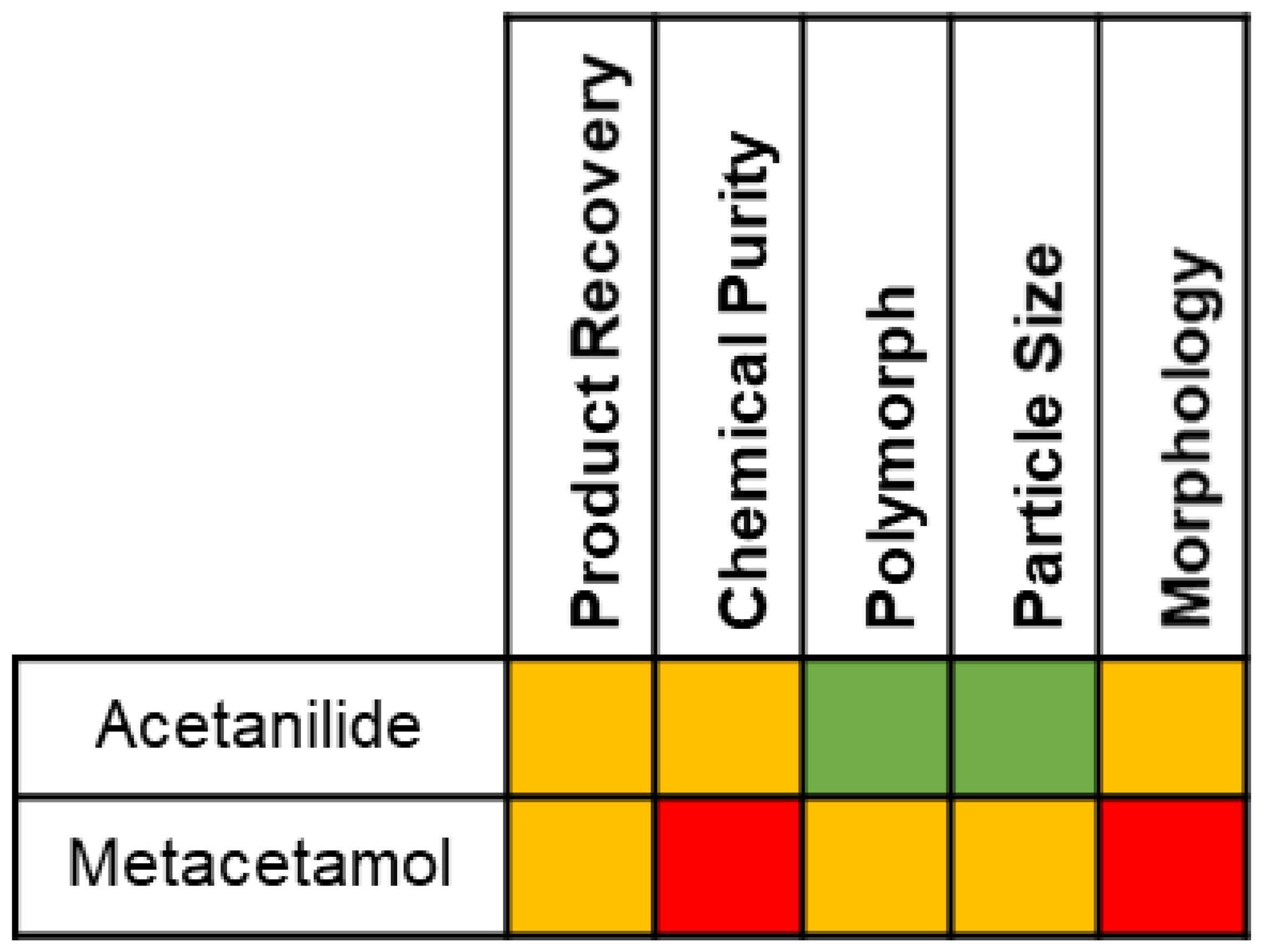
Effect of Impurities
3.2. Analysis of Product Attributes
3.2.1. Crystal Purity
3.2.2. Polymorphic Form
 ) on Figure 3). Suspension of these Form II crystals in 2-propanol saturated with paracetamol further increased the purity to 99.2 mol% (denoted ( ) in Figure 4), while XRPD indicates this slurry process also induced a polymorphic transformation to stable paracetamol Form I (Figure 6d).
) on Figure 3). Suspension of these Form II crystals in 2-propanol saturated with paracetamol further increased the purity to 99.2 mol% (denoted ( ) in Figure 4), while XRPD indicates this slurry process also induced a polymorphic transformation to stable paracetamol Form I (Figure 6d).3.2.3. Particle Size
, Figure 10a). The span of the distributions varies across the series (Table S4). Across the metacetamol-contaminated samples, the average span of the crystallization product samples is 1.18 (standard deviation = 0.50). At the extreme of this range was the case of Xi,p = 6.78 mol%, where the span of the distribution is 2.19. We hypothesise that, as these particles are very fine needles (Figure 8f), breakage occurs during the sample dispersion for measurement, which results in a bi-modal particle size distribution (Figure S8f) and hence a large value for the span.3.2.4. Crystal Morphology
, Figure 11a). It is noted here that crystals with the highest Xi,F (6.78 mol%) of metacetamol were very fragile needles (Figure 8f) and were damaged during the compressed air dispersion required for particle-shape distribution analysis, even when low energy dispersion techniques were used, and so the aspect ratio value obtained for this sample may be substantially higher than the actual one.4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ter Horst, J.H.; Schmidt, C.; Ulrich, J. Handbook of Crystal Growth, 2nd ed.; Elsevier, B.V.: Amsterdam, The Netherlands, 2015; pp. 1317–1349. [Google Scholar]
- European Medicines Agency. Impurities in New Drug Products (Q3B(R2)). In Proceeding of International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals from Human Use. Available online: https://www.ema.europa.eu/en/ich-q3b-r2-impurities-new-drug-products (accessed on 20 September 2021).
- Urwin, S.J.; Levilain, G.; Marziano, I.; Merritt, J.M.; Houson, I.; Ter Horst, J.H. A Structured Approach to Cope with Impurities during Industrial Crystallization Development. Org. Process. Res. Dev. 2020, 24, 1443–1456. [Google Scholar] [CrossRef] [PubMed]
- Moynihan, H.A.; Horgan, D.E. Impurity Occurrence and Removal in Crystalline Products from Process Reactions. Org. Process. Res. Dev. 2017, 21, 689–704. [Google Scholar] [CrossRef]
- Darmali, C.; Mansouri, S.; Yazdanpanah, N.; Woo, M.W. Mechanisms and Control of Impurities in Continuous Crystallization: A Review. Ind. Eng. Chem. Res. 2018, 58, 1463–1479. [Google Scholar] [CrossRef]
- Nguyen, T.T.H.; Khan, A.; Bruce, L.M.; Forbes, C.; O’Leary, R.L.; Price, C.J. The Effect of Ultrasound on the Crystallization of Paracetamol in the Presence of Structurally Similar Impurities. Crystals 2017, 7, 294. [Google Scholar] [CrossRef] [Green Version]
- Hendriksen, B.A.; Grant, D.J.W.; Meenan, P.; Green, D.A. Crystallization of paracetamol (acetaminophen) in the presence of structurally related substances. J. Cryst. Growth 1998, 183, 629–640. [Google Scholar] [CrossRef]
- Lee, A.Y.; Erdemir, D.; Myerson, A.S. Crystal Polymorphism in Chemical Process Development. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 259–280. [Google Scholar] [CrossRef] [PubMed]
- Agnew, L.R.; Cruickshank, D.L.; McGlone, T.; Wilson, C.C. Controlled production of the elusive metastable form II of acetaminophen (paracetamol): A fully scalable templating approach in a cooling environment. Chem. Commun. 2016, 52, 7368–7371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, G.; Wu, Y.; Yang, X.; Duan, X.; Zhou, X. Effect of polymorphism on the purity of l-glutamic acid. J. Cryst. Growth 2013, 373, 78–81. [Google Scholar] [CrossRef]
- Tanoury, G.J.; Hett, R.; Kessler, D.W.; Wald, S.A.; Senanayake, C.H. Taking Advantage of Polymorphism To Effect an Impurity Removal: Development of a Thermodynamic Crystal Form of (R,R)-Formoterol Tartrate. Org. Process. Res. Dev. 2002, 6, 855–862. [Google Scholar] [CrossRef]
- Black, S.; Cuthbert, M.W.; Roberts, R.J.; Stensland, B. Increased Chemical Purity Using a Hydrate. Cryst. Growth Des. 2004, 4, 539–544. [Google Scholar] [CrossRef]
- Ter Horst, J.H.; van Rosmalen, R.M.; Geertman, R.M. Additives: Molecular Design. In Encyclopedia of Separation Science; Academic Press: Cambridge, MA, USA, 2000; pp. 931–940. [Google Scholar]
- Gu, C.; Grant, D.J.W. Relationship Between Particle Size and Impurity Incorporation During Crystallization of (+)-Pseudoephedrine Hydrochloride, Acetaminophen, and Adipic Acid from Aqueous Solution. Pharm. Res. 2002, 19, 1068–1070. [Google Scholar] [CrossRef] [PubMed]
- Steendam, R.R.E.; Keshavarz, L.; de Souza, B.P.; Frawley, P.J. Thermodynamic properties of paracetamol impurities 4-nitrophenol and 4″-chloroacetanilide and the impact of such impurities on the crystallization of paracetamol from solution. J. Chem. Thermodyn. 2019, 133, 85–92. [Google Scholar] [CrossRef]
- MacLeod, C.S.; Muller, F. On the Fracture of Pharmaceutical Needle-Shaped Crystals during Pressure Filtration: Case Studies and Mechanistic Understanding. Org. Process. Res. Dev. 2012, 16, 425–434. [Google Scholar] [CrossRef]
- Thomas, L.H.; Wales, C.; Zhao, L.; Wilson, C.C. Paracetamol Form II: An Elusive Polymorph through Facile Multicomponent Crystallization Routes. Cryst. Growth Des. 2011, 11, 1450–1452. [Google Scholar] [CrossRef]
- Agnew, L.R.; McGlone, T.; Wheatcroft, H.P.; Robertson, A.; Parsons, A.R.; Wilson, C.C. Continuous Crystallization of Paracetamol (Acetaminophen) Form II: Selective Access to a Metastable Solid Form. Cryst. Growth Des. 2017, 17, 2418–2427. [Google Scholar] [CrossRef]
- Gaisford, S.; Buanz, A.B.; Jethwa, N. Characterisation of paracetamol form III with rapid-heating DSC. J. Pharm. Biomed. Anal. 2010, 53, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Telford, R.; Seaton, C.C.; Clout, A.; Buanz, A.; Gaisford, S.; Williams, G.R.; Prior, T.J.; Okoye, C.H.; Munshi, T.; Scowen, I.J. Stabilisation of metastable polymorphs: The case of paracetamol form III. Chem. Commun. 2016, 52, 12028–12031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, G.; Frampton, C.S. Physicochemical Characterization of the Orthorhombic Polymorph of Paracetamol Crystallized from Solution. J. Pharm. Sci. 1998, 87, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Tang, S.K.; Hao, H.; Davey, R.J.; Vetter, T. Quantifying the Inherent Uncertainty Associated with Nucleation Rates Estimated from Induction Time Data Measured in Small Volumes. Cryst. Growth Des. 2017, 17, 2852–2863. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; ter Horst, J.H. Crystal Nucleation Rates from Probability Distributions of Induction Times. Cryst. Growth Des. 2010, 11, 256–261. [Google Scholar] [CrossRef]
- Kulkarni, S.A.; Kadam, S.S.; Meekes, H.; Stankiewicz, A.I.; ter Horst, J.H. Crystal Nucleation Kinetics from Induction Times and Metastable Zone Widths. Cryst. Growth Des. 2013, 13, 2435–2440. [Google Scholar] [CrossRef]
- De Wet, F.N.; Gerber, J.J.; Lötter, A.P.; Van der Watt, J.G.; Dekker, T.G. A study of the changes during heating of paracetamol. Drug Dev. Ind. Pharm. 1998, 24, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Granberg, R.A.; Rasmuson, C. Solubility of Paracetamol in Pure Solvents. J. Chem. Eng. Data 1999, 44, 1391–1395. [Google Scholar] [CrossRef]
- Rycerz, L. Practical remarks concerning phase diagrams determination on the basis of differential scanning calorimetry measurements. J. Therm. Anal. Calorim. 2013, 113, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.J. McGlone, T. Yerdelen, S. Srirambhatla, V. Mabbott, F. Gurung, R. Briuglia, M.L. Ahmed, B. Polyzois, H. McGinty, J.; et al. Enabling precision manufacturing of active pharmaceutical ingredients: Workflow for seeded cooling continuous crystallizations. Mol. Syst. Des. Eng. 2018, 3, 518–549. [Google Scholar] [CrossRef] [Green Version]
- Keshavarz, L.; Steendam, R.R.E.; Blijlevens, M.A.R.; Pishnamazi, M.; Frawley, P.J. Influence of Impurities on the Solubility, Nucleation, Crystallization, and Compressibility of Paracetamol. Cryst. Growth Des. 2019, 19, 4193–4201. [Google Scholar] [CrossRef]
- Hendriksen, B.A.; Grant, D.J. The effect of structurally related substances on the nucleation kinetics of paracetamol (acetaminophen). J. Cryst. Growth 1995, 156, 252–260. [Google Scholar] [CrossRef]
- Saleemi, A.; Onyemelukwe, I.I.; Nagy, Z. Effects of a structurally related substance on the crystallization of paracetamol. Front. Chem. Sci. Eng. 2013, 7, 79–87. [Google Scholar] [CrossRef]
- Liu, Y.; Gabriele, B.; Davey, R.J.; Cruz-Cabeza, A.J. Concerning Elusive Crystal Forms: The Case of Paracetamol. J. Am. Chem. Soc. 2020, 142, 6682–6689. [Google Scholar] [CrossRef]
- Perrin, M.-A.; Neumann, M.A.; Elmaleh, H.; Zaske, L. Crystal structure determination of the elusive paracetamol Form III. Chem. Commun. 2009, 22, 3181–3183. [Google Scholar] [CrossRef]
- Haisa, M.; Kashino, S.; Maeda, H. The orthorhombic form of p-hydroxyacetanilide. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1974, 30, 2510–2512. [Google Scholar] [CrossRef]
- Cesar, M.A.B.; Ng, K.M. Improving Product Recovery in Fractional Crystallization Processes: Retrofit of an Adipic Acid Plant. Ind. Eng. Chem. Res. 1999, 38, 823–832. [Google Scholar] [CrossRef]
- Alvarez, A.J.; Singh, A.; Myerson, A.S. Crystallization of Cyclosporine in a Multistage Continuous MSMPR Crystallizer. Cryst. Growth Des. 2011, 11, 4392–4400. [Google Scholar] [CrossRef]
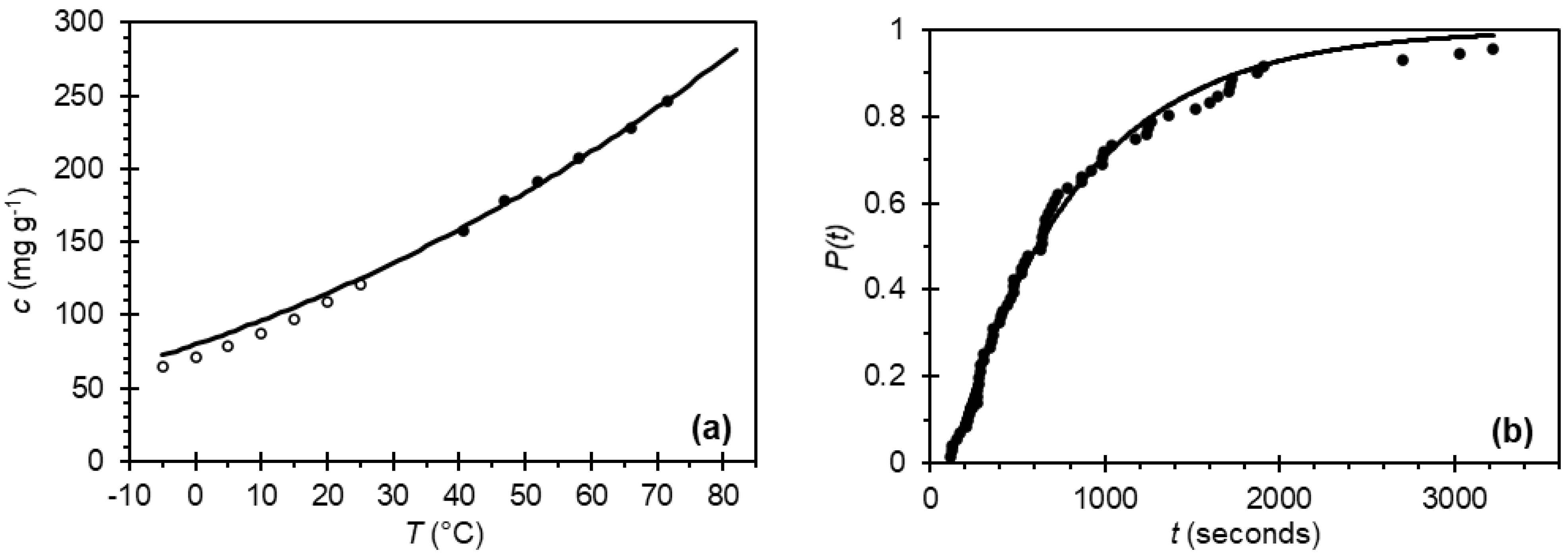
 ) and compared with published gravimetric values (
) and compared with published gravimetric values (  ). (b) Probability distribution of induction times for paracetamol in 2-propanol at a supersaturation ratio S = 2.10 resulting in a growth time tg = 24 ± 4 s, a nucleation rate J = 1376 ± 17 m−3 s−1 and median induction time tind = 480 s.
) and compared with published gravimetric values ( ). (b) Probability distribution of induction times for paracetamol in 2-propanol at a supersaturation ratio S = 2.10 resulting in a growth time tg = 24 ± 4 s, a nucleation rate J = 1376 ± 17 m−3 s−1 and median induction time tind = 480 s.
). (b) Probability distribution of induction times for paracetamol in 2-propanol at a supersaturation ratio S = 2.10 resulting in a growth time tg = 24 ± 4 s, a nucleation rate J = 1376 ± 17 m−3 s−1 and median induction time tind = 480 s.
) and compared with published gravimetric values ( ). (b) Probability distribution of induction times for paracetamol in 2-propanol at a supersaturation ratio S = 2.10 resulting in a growth time tg = 24 ± 4 s, a nucleation rate J = 1376 ± 17 m−3 s−1 and median induction time tind = 480 s.
 ) Paracetamol in presence of metacetamol, linear regression Xp = −0.735 Xi,F + 100.39, R2 = 0.998. (
) Paracetamol in presence of metacetamol, linear regression Xp = −0.735 Xi,F + 100.39, R2 = 0.998. (  ) Paracetamol in the presence of acetanilide, linear regression Xp = −0.141 Xi,F + 99.9, R2 = 0.999. ( ) Paracetamol in presence of metacetamol where Form II was crystallized. (
) Paracetamol in the presence of acetanilide, linear regression Xp = −0.141 Xi,F + 99.9, R2 = 0.999. ( ) Paracetamol in presence of metacetamol where Form II was crystallized. (  ) denotes the specification of 99.5 mol% product purity. All experimental solutions nucleated at a supersaturation S = 2.10. Error bars denote the standard error.
) Paracetamol in presence of metacetamol, linear regression Xp = −0.735 Xi,F + 100.39, R2 = 0.998. ( ) Paracetamol in the presence of acetanilide, linear regression Xp = −0.141 Xi,F + 99.9, R2 = 0.999. ( ) Paracetamol in presence of metacetamol where Form II was crystallized. ( ) denotes the specification of 99.5 mol% product purity. All experimental solutions nucleated at a supersaturation S = 2.10. Error bars denote the standard error.
) denotes the specification of 99.5 mol% product purity. All experimental solutions nucleated at a supersaturation S = 2.10. Error bars denote the standard error.
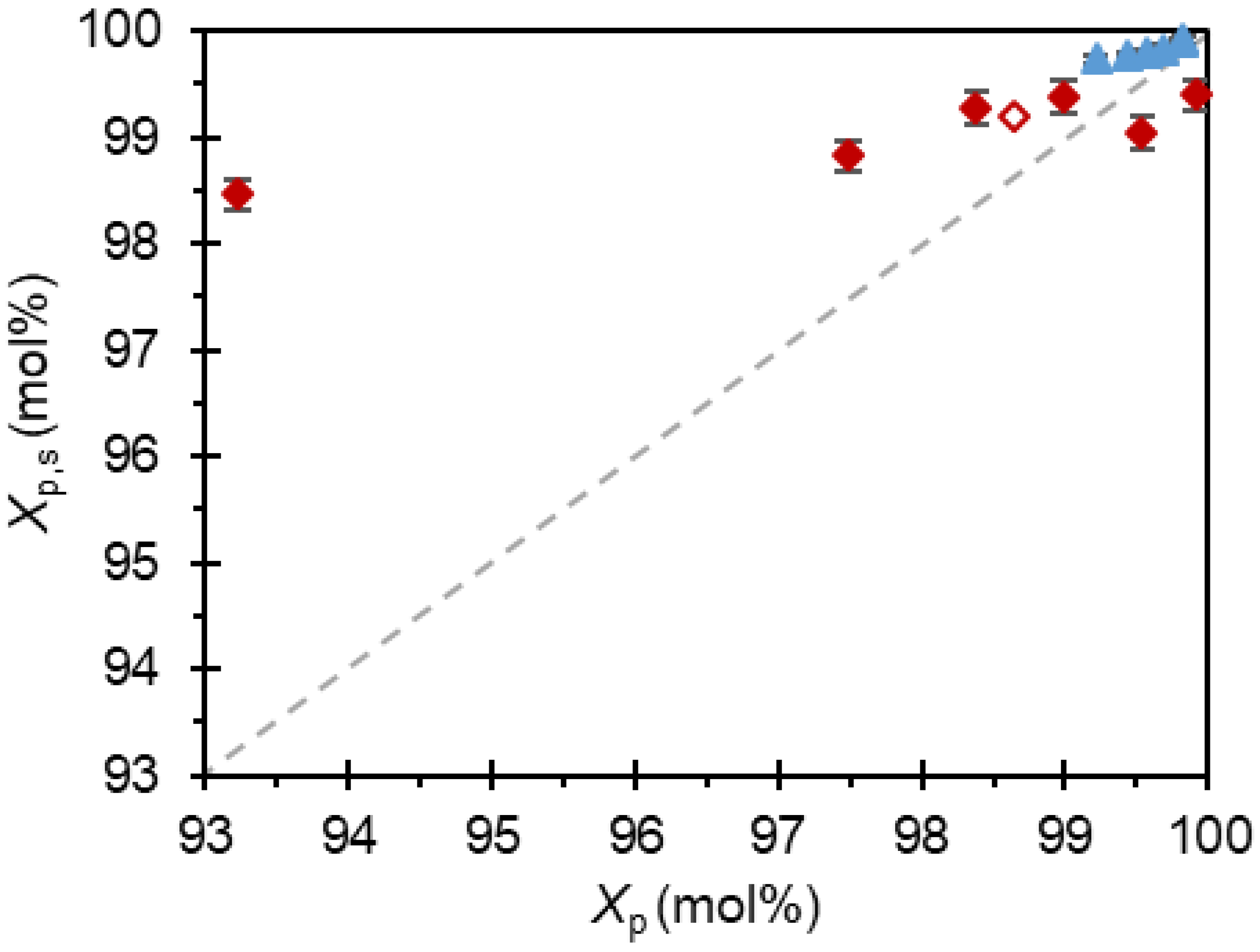
) Paracetamol in presence of metacetamol, linear regression Xp = −0.735 Xi,F + 100.39, R2 = 0.998. ( ) Paracetamol in the presence of acetanilide, linear regression Xp = −0.141 Xi,F + 99.9, R2 = 0.999. ( ) Paracetamol in presence of metacetamol where Form II was crystallized. ( ) denotes the specification of 99.5 mol% product purity. All experimental solutions nucleated at a supersaturation S = 2.10. Error bars denote the standard error. ) Paracetamol in presence of metacetamol. ( ) Paracetamol in the presence of acetanilide. ( ) Paracetamol in presence of metacetamol where Form II was crystallized. The diagonal line (
) Paracetamol in presence of metacetamol. ( ) Paracetamol in the presence of acetanilide. ( ) Paracetamol in presence of metacetamol where Form II was crystallized. The diagonal line (  ) is added for comparison of purifications. Error bars denote standard error.
) Paracetamol in presence of metacetamol. ( ) Paracetamol in the presence of acetanilide. ( ) Paracetamol in presence of metacetamol where Form II was crystallized. The diagonal line ( ) is added for comparison of purifications. Error bars denote standard error.
) is added for comparison of purifications. Error bars denote standard error.
) Paracetamol in presence of metacetamol. ( ) Paracetamol in the presence of acetanilide. ( ) Paracetamol in presence of metacetamol where Form II was crystallized. The diagonal line ( ) is added for comparison of purifications. Error bars denote standard error.

 ) a-axis, (
) a-axis, (  ) b-axis and ( ) c-axis. (b) Acetanilide ( ) a-axis, (
) b-axis and ( ) c-axis. (b) Acetanilide ( ) a-axis, (  ) b-axis and (
) b-axis and (  ) c-axis. Reference structure for paracetamol was used from the CCDC to determine length deviation. Error bars denote standard error.
) a-axis, ( ) b-axis and ( ) c-axis. (b) Acetanilide ( ) a-axis, ( ) b-axis and ( ) c-axis. Reference structure for paracetamol was used from the CCDC to determine length deviation. Error bars denote standard error.
) c-axis. Reference structure for paracetamol was used from the CCDC to determine length deviation. Error bars denote standard error.
) a-axis, ( ) b-axis and ( ) c-axis. (b) Acetanilide ( ) a-axis, ( ) b-axis and ( ) c-axis. Reference structure for paracetamol was used from the CCDC to determine length deviation. Error bars denote standard error.

 ) paracetamol in absence of impurities, ( ) paracetamol contaminated with metacetamol ( ) paracetamol Form II contaminated with metacetamol and ( ) paracetamol contaminated with acetanilide. (a) Crystallization product. (b) Reslurry product.
) paracetamol in absence of impurities, ( ) paracetamol contaminated with metacetamol ( ) paracetamol Form II contaminated with metacetamol and ( ) paracetamol contaminated with acetanilide. (a) Crystallization product. (b) Reslurry product.
) paracetamol in absence of impurities, ( ) paracetamol contaminated with metacetamol ( ) paracetamol Form II contaminated with metacetamol and ( ) paracetamol contaminated with acetanilide. (a) Crystallization product. (b) Reslurry product.
) paracetamol in absence of impurities, ( ) paracetamol contaminated with metacetamol ( ) paracetamol Form II contaminated with metacetamol and ( ) paracetamol contaminated with acetanilide. (a) Crystallization product. (b) Reslurry product. ) Paracetamol in absence of impurities, ( ) paracetamol contaminated with metacetamol ( ) paracetamol Form II (Form I after slurrying) contaminated with metacetamol and ( ) paracetamol contaminated with acetanilide.
) Paracetamol in absence of impurities, ( ) paracetamol contaminated with metacetamol ( ) paracetamol Form II (Form I after slurrying) contaminated with metacetamol and ( ) paracetamol contaminated with acetanilide.
) Paracetamol in absence of impurities, ( ) paracetamol contaminated with metacetamol ( ) paracetamol Form II (Form I after slurrying) contaminated with metacetamol and ( ) paracetamol contaminated with acetanilide.
) Paracetamol in absence of impurities, ( ) paracetamol contaminated with metacetamol ( ) paracetamol Form II (Form I after slurrying) contaminated with metacetamol and ( ) paracetamol contaminated with acetanilide.
 ), through moderate effect (
), through moderate effect (  ) to significant negative effect (
) to significant negative effect (  ).
), through moderate effect ( ) to significant negative effect ( ).
).
), through moderate effect ( ) to significant negative effect ( ).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urwin, S.J.; Yerdelen, S.; Houson, I.; ter Horst, J.H. Impact of Impurities on Crystallization and Product Quality: A Case Study with Paracetamol. Crystals 2021, 11, 1344. https://doi.org/10.3390/cryst11111344
Urwin SJ, Yerdelen S, Houson I, ter Horst JH. Impact of Impurities on Crystallization and Product Quality: A Case Study with Paracetamol. Crystals. 2021; 11(11):1344. https://doi.org/10.3390/cryst11111344
Chicago/Turabian StyleUrwin, Stephanie J., Stephanie Yerdelen, Ian Houson, and Joop H. ter Horst. 2021. "Impact of Impurities on Crystallization and Product Quality: A Case Study with Paracetamol" Crystals 11, no. 11: 1344. https://doi.org/10.3390/cryst11111344
APA StyleUrwin, S. J., Yerdelen, S., Houson, I., & ter Horst, J. H. (2021). Impact of Impurities on Crystallization and Product Quality: A Case Study with Paracetamol. Crystals, 11(11), 1344. https://doi.org/10.3390/cryst11111344