
Co-Deposition Mechanisms of Calcium Sulfate and Calcium Carbonate Scale in Produced Water


Abstract
:1. Introduction
2. Background Theory
2.1. Crystallization
- (1)
- (2)
- At a higher supersaturation, the ion pair concentration increases and aggregates to form larger particles. In this process, the aggregation is in a dynamic equilibrium state of dissolution and aggregation in the solution [1]. The polyelectrolyte can be adsorbed on the surface of the molecular aggregation, affecting its growth and dissolution kinetics [17].
- (3)
- When the aggregate length reaches the critical size, determined by the supersaturation of the solution, these larger particles will no longer continue to dissolve, and the solid particles begin to nucleate [37]. At this stage, the nucleation of the main particles in solution occurs by homogeneous nucleation, while that of aggregates adsorbed on a surface occurs by heterogeneous nucleation. These two nucleation processes may exist at the interface of heterogeneous nucleation, and it is difficult to distinguish the specific nucleation sites (typically dust, impurities, or other material surfaces) of a substance [38]. Once the particles achieve nucleation, there are several potential sediment growth mechanisms, either in the macroscopic surface or in the solution.
- (4)
- The crystal nucleus continues to grow and aggregate in the supersaturated solution. At the same time, the particles continue to grow on the surface of the scale layer after nucleation, and finally scaling occurs.
2.2. Calcium Sulfate and Calcium Carbonate
2.3. Previous Work on Co-Precipitation
- (1)
- (2)
3. Experimental Methods and Materials
3.1. Materials
3.2. Solution Preparation
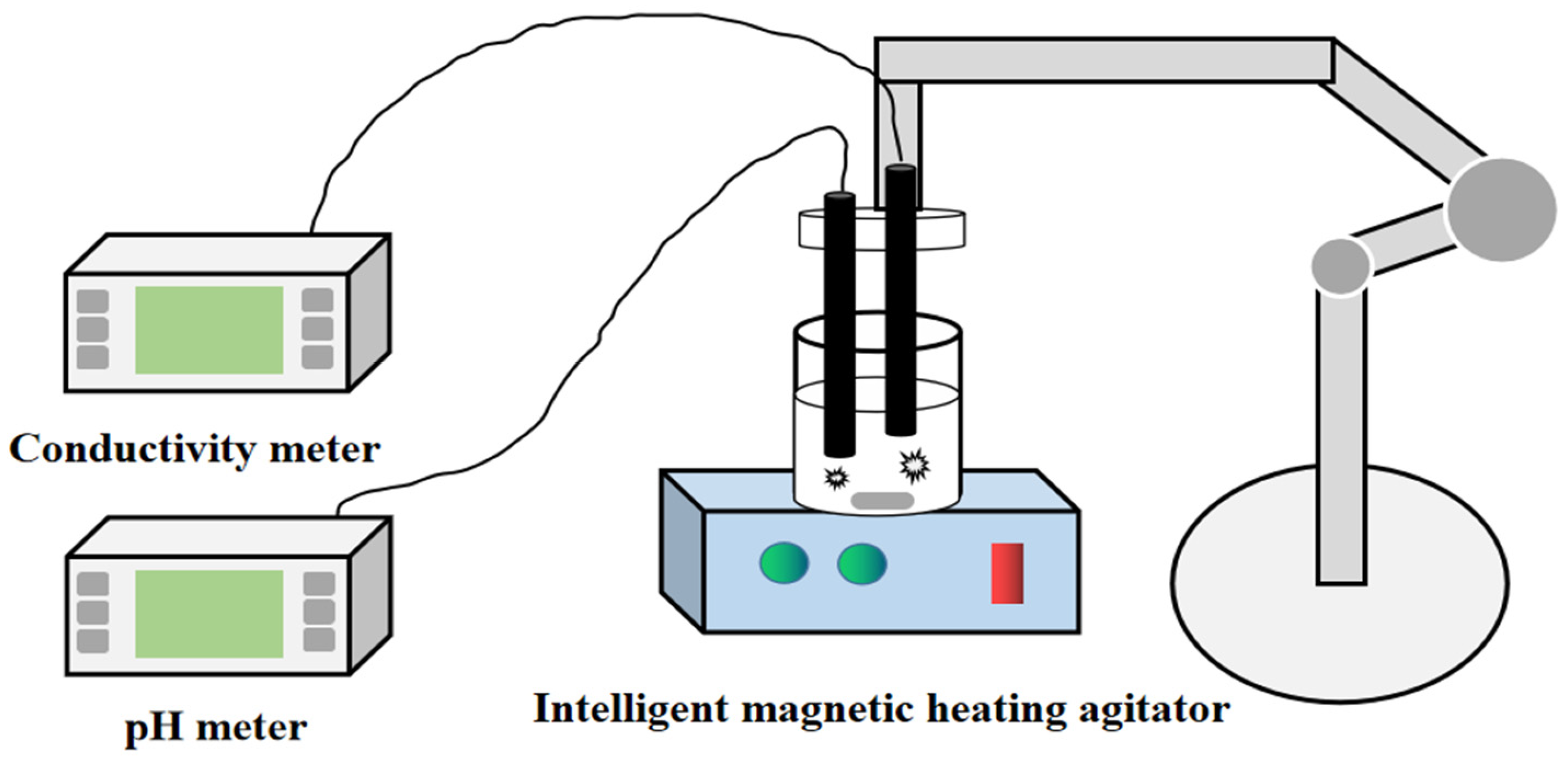
3.3. Experiments and Analysis Methods
3.3.1. Determination of Conductivity and pH Value
3.3.2. Microscopic Observation
3.3.3. Analysis by XRD
4. Results and Discussion
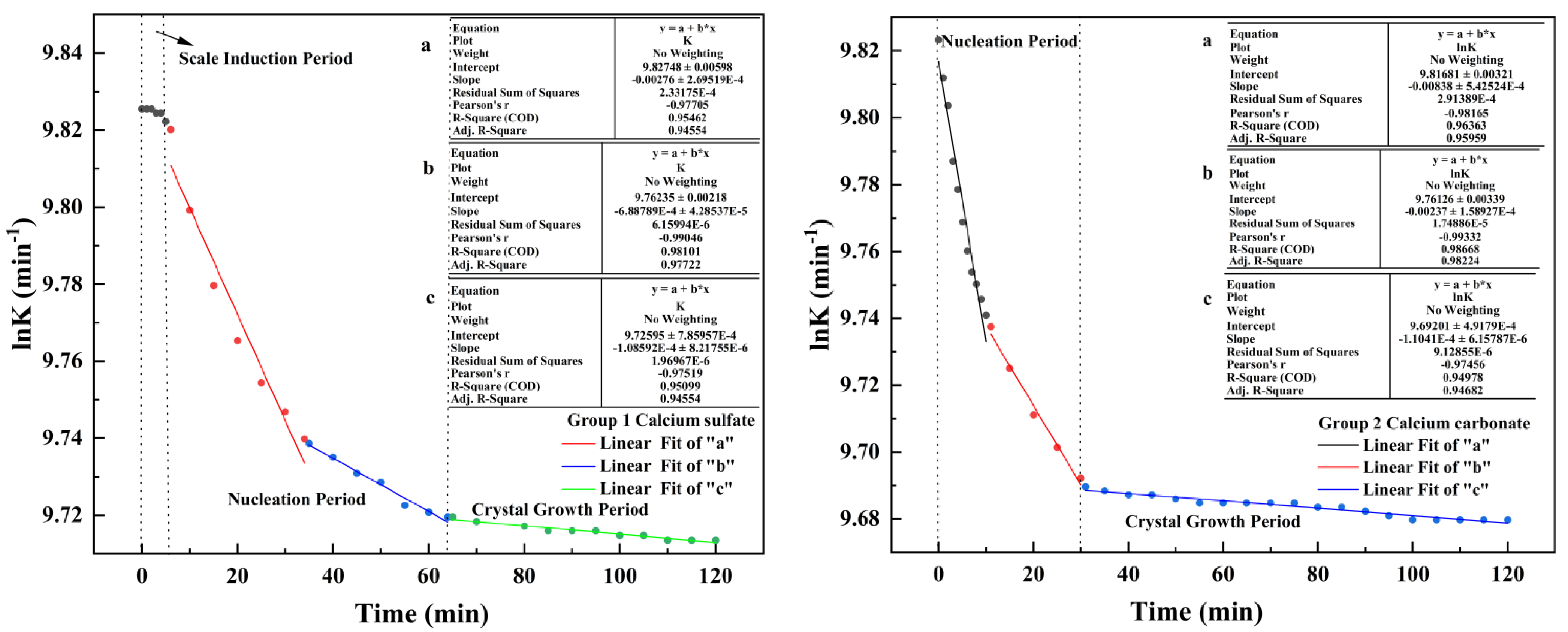
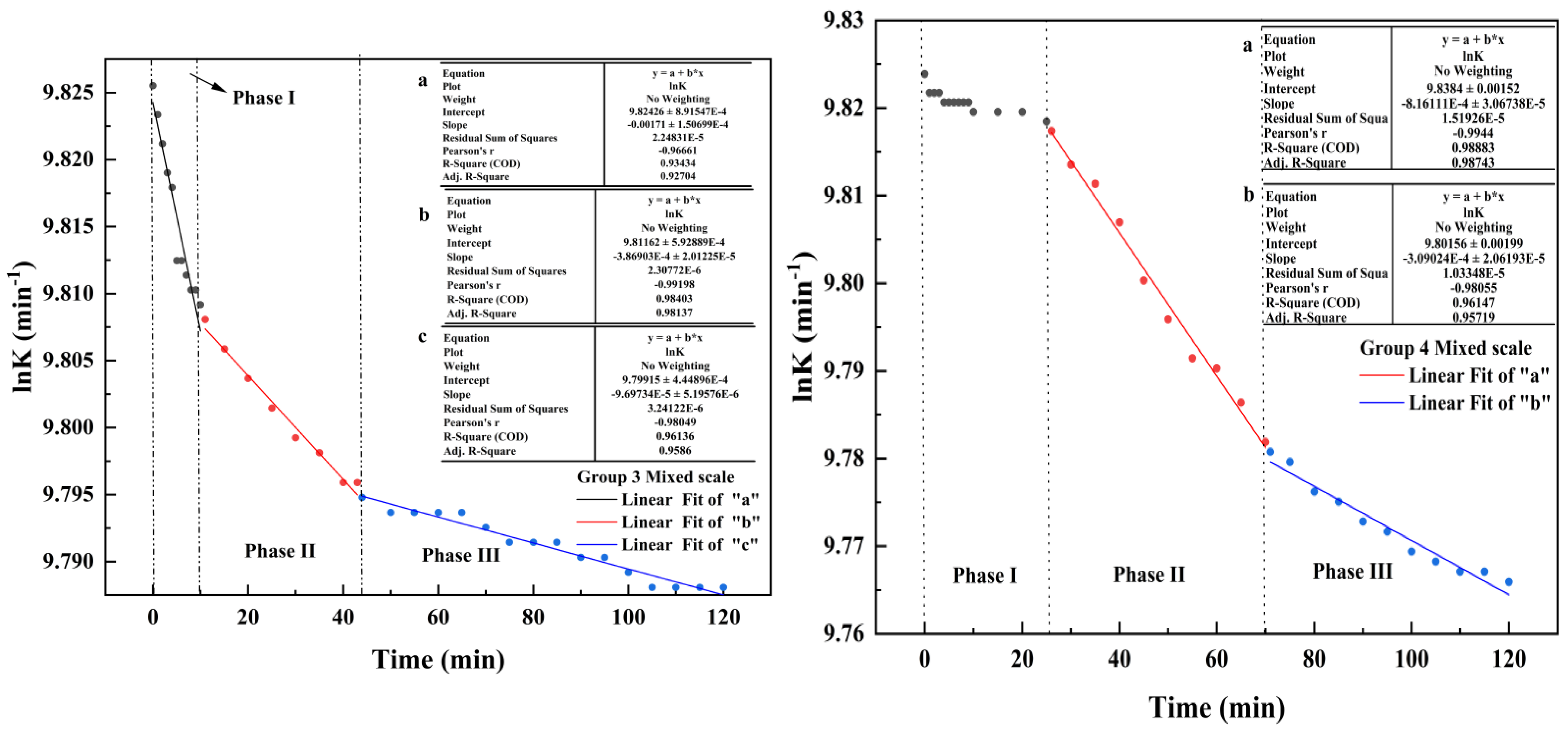
4.1. Changes in Electrical Conductivity during Scale Deposition
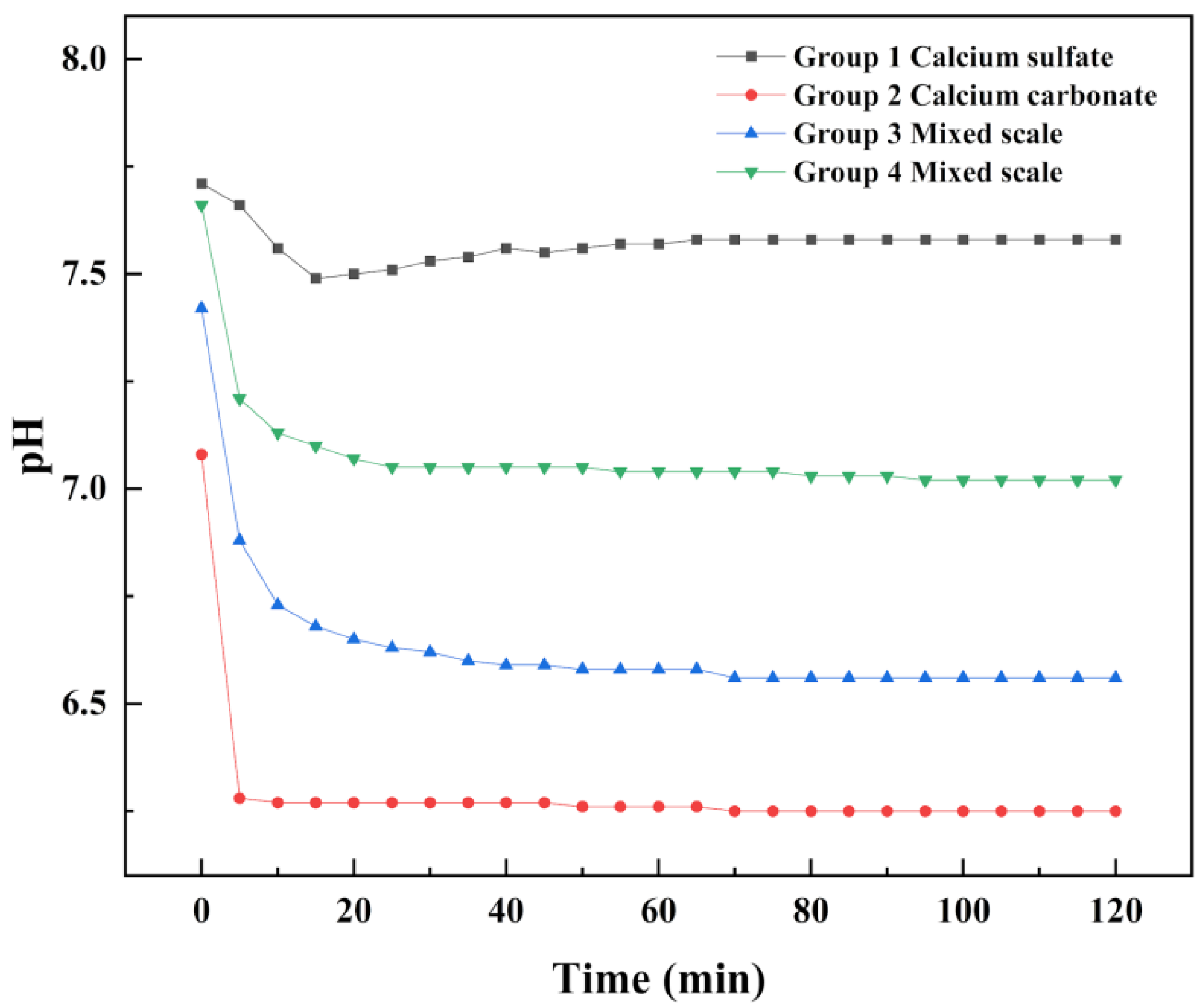
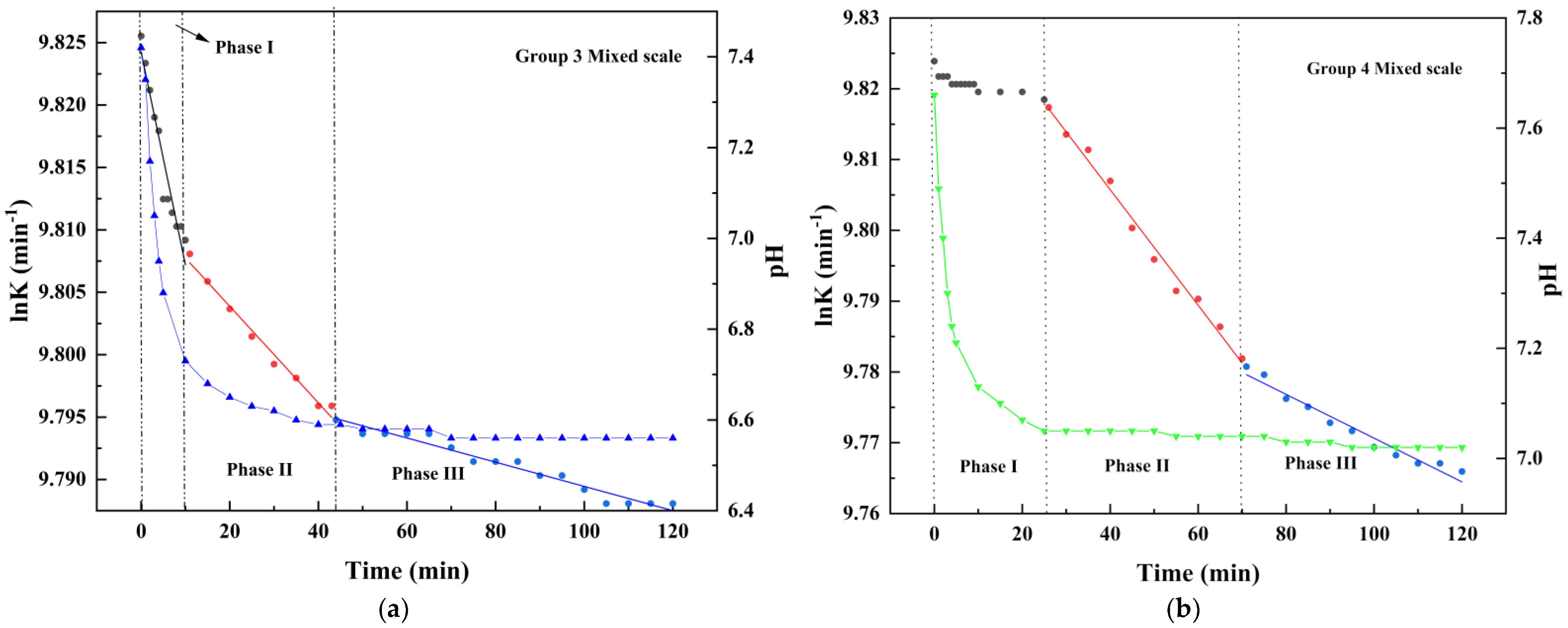
4.2. Changes of pH Value in Scale Deposition Process
4.3. Changes of Crystal Morphology in Scale Deposition Process
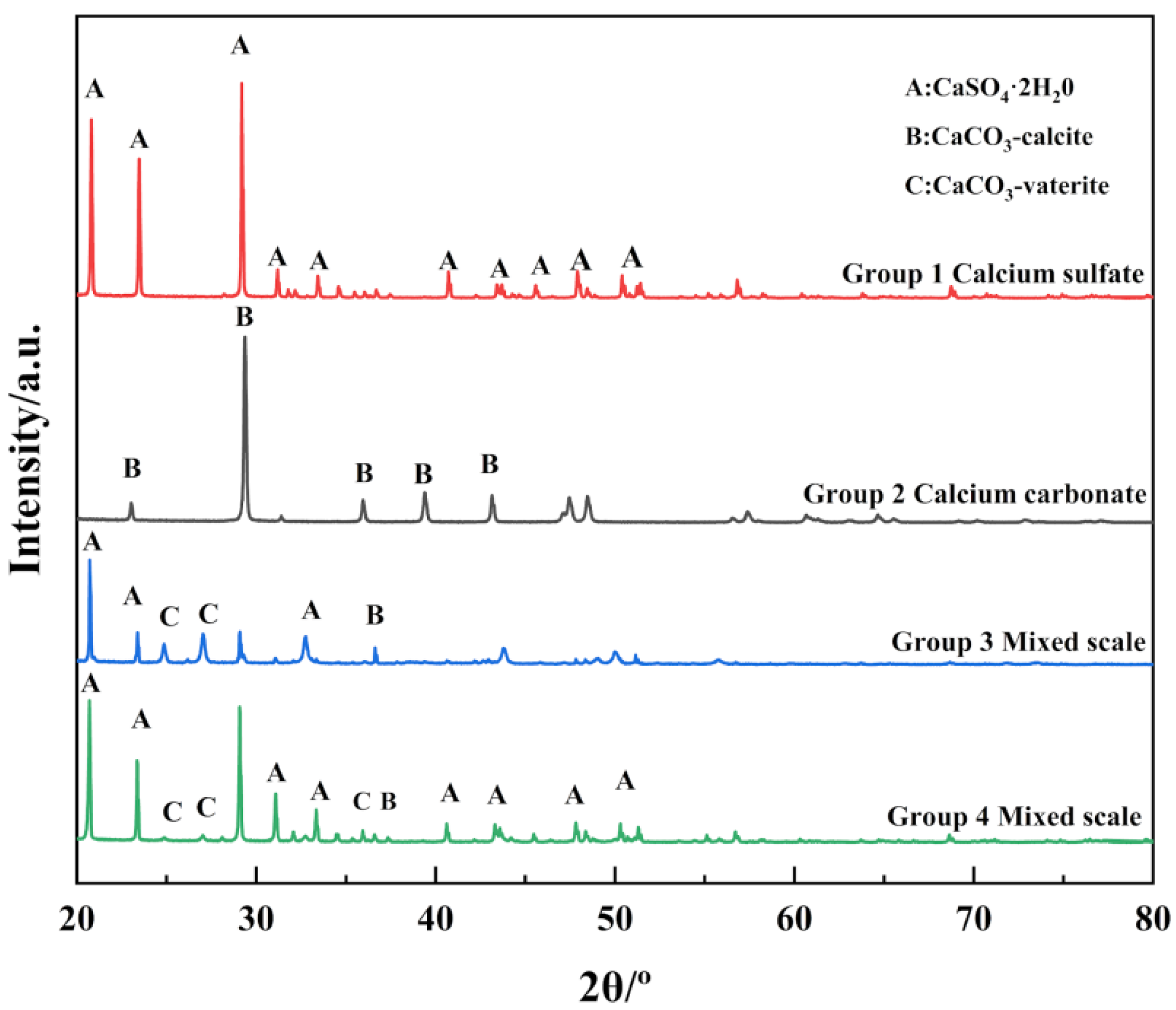
4.4. XRD Analysis of Scale Samples
5. CaCO3-CaSO4 Co-Deposition Model
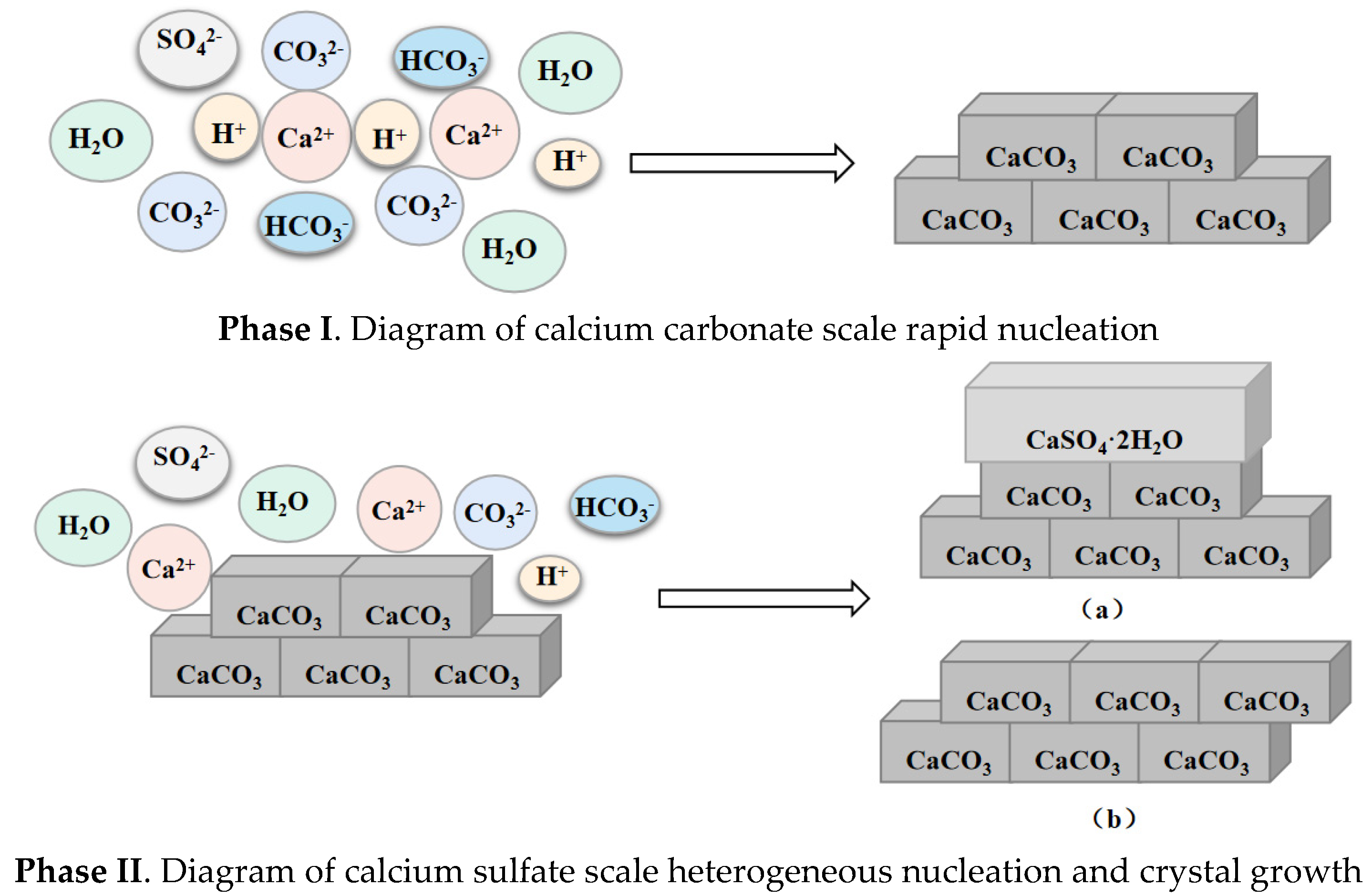
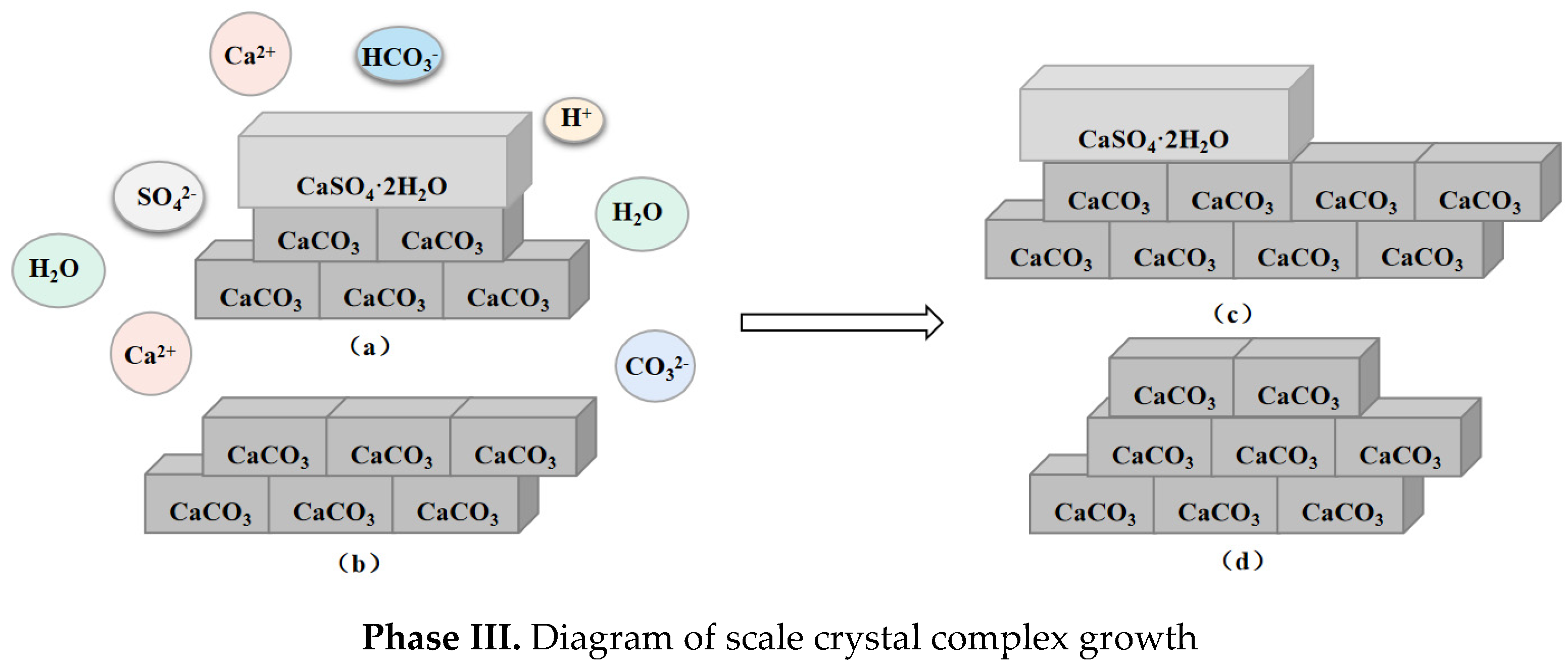
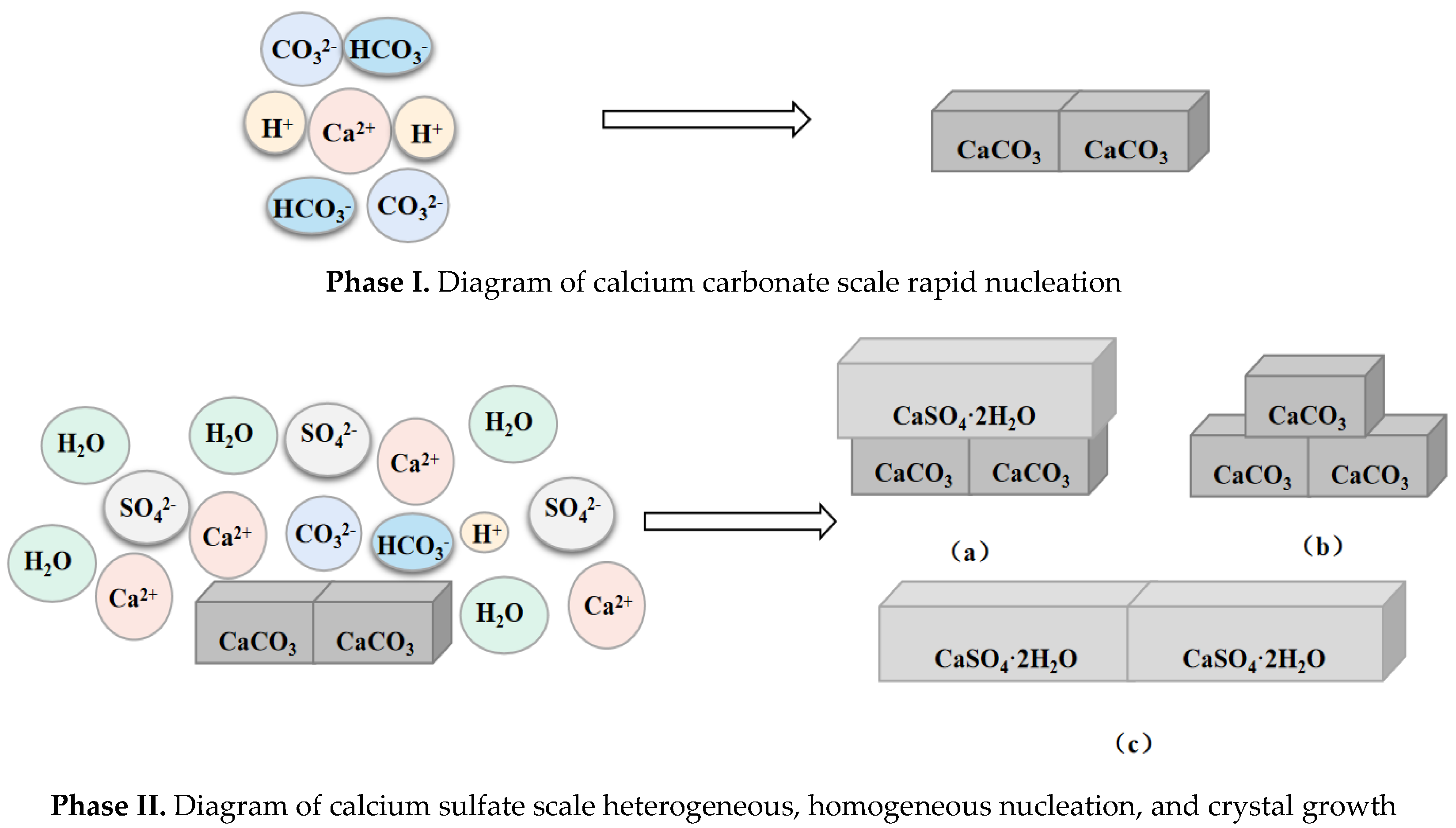
5.1. Calcium Carbonate as Dominant Scale Type during Co-Deposition
5.2. Calcium Sulfate as Dominant Scale Type during Co-Deposition
6. Conclusions
- (1)
- In this paper, four groups of mixed solutions with equal volume, equal Ca2+ content, and anion content that can react completely with Ca2+ ions (ideal state) were studied. Obvious scaling induction, nucleation, and crystal growth periods in pure calcium sulfate scale deposition. While pure calcium carbonate scale deposition, rapid scale formation, and the scale induction period was not obvious. In the process of mixed scale co-deposition, calcium carbonate scale would take the lead in the formation, and the content of HCO3− increases, which prolonged the induction period of calcium sulfate scale, slowed the growth of calcium sulfate scale, and inhibited its growth process.
- (2)
- Compared with the co-deposition products containing calcium carbonate, the adhesion of pure calcium sulfate deposits was much lower. In the presence of calcium sulfate, calcium carbonate scales which were usually very adhesive and tough, lost their strength and became less adhesive. The adhesion of co-deposition products was between the calcium sulfate scale and calcium carbonate scale.
- (3)
- Calcium sulfate scale in the co-deposition and pure material deposition, no significant difference in the crystalline form, gypsum form. The scale of calcium carbonate in co-deposition is different from those in pure material deposition. In pure material deposition, the calcium carbonate scale deposition form was mostly stable calcite form. When co-deposition occurs, SO42− might occupy the active sites on the crystal surface, inhibit the crystal growth of the calcium carbonate scale, and make it difficult to transform from vaterite to calcite.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abdel-Aal, N.; Satoh, K.; Sawada, K. Study of Adhesion Mechanism of Calcareous Scaling by Using Quartz Crystal Microbalance Technique. Anal. Sci. Suppl. 2007, 17, i825–i828. [Google Scholar]
- Lioliou, M.G.; Paraskeva, C.A.; Koutsoukos, P.G.; Payatakes, A.C. Heterogeneous nucleation and growth of calcium carbonate on calcite and quartz. J. Colloid Interface Sci. 2007, 308, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.M.; Fu, W.; Bai, H.T.; Zhou, L.; Ma, Y. Study on Compatibility between Formation Water and Injection Water in Jiyuan Oilfield. Oilfield Chem. 2012. Available online: https://en.cnki.com.cn/Article_en/CJFDTotal-YJHX201201008.htm (accessed on 20 November 2021).
- Bukuaghangin, O.; Sanni, O.; Kapur, N.; Huggan, M.; Neville, A.; Charpentier, T. Kinetics study of barium sulphate surface scaling and inhibition with a once-through flow system. J. Pet. Sci. Eng. 2016, 147, 699–706. [Google Scholar] [CrossRef] [Green Version]
- Al-Amrie, O.; Peltier, S.; Pearce, A.; Abu-Dhabi, T.; Al-Yafei, A.; Morel, D.; Bourrel, M.; Bursaux, R.; Cordelier, P.; Jouenne, S.; et al. The first successful chemical EOR pilot in the UAE: One spot pilot in high temperature, high salinity carbonate reservoir. In Proceedings of the Abu Dhabi International Petroleum Exhibition and Conference, Abu Dhabi, United Arab Emirates, 15–18 November 2015. [Google Scholar]
- Bodin, H.; Persson, J.; Englund, J.E.; Milberg, P. Influence of residence time analyses on estimates of wetland hydraulics and pollutant removal. J. Hydrol. 2013, 501, 1–12. [Google Scholar] [CrossRef]
- Naseri, S.; Moghadasi, J.; Jamialahmadi, M. Effect of temperature and calcium ion concentration on permeability reduction due to composite barium and calcium sulfate precipitation in porous media. J. Nat. Gas Sci. Eng. 2015, 22, 299–312. [Google Scholar] [CrossRef]
- Ramstad, K.; Tydal, T.; Askvik, K.M.; Fotland, P. Predicting Carbonate Scale in Oil Producers from High-Temperature Reservoirs. SPE J. 2005, 10, 363–373. [Google Scholar] [CrossRef]
- Moghadasi, J.; Jamialahmadi, M.; Müller-Steinhagen, H.; Sharif, A.; Ghalambor, A.; Izadpanah, M.R.; Motaie, E. Scale formation in Iranian oil reservoir and production equipment during water injection. In Proceedings of the International Symposium on Oilfield Scale, Aberdeen, UK, 29–30 January 2003. [Google Scholar]
- Tung, N.P.; Phuong, N.T.P.; Long, B.Q.K.; Duy, N.H. Scale Inhibitors for Co-Deposited Calcium Sulfate and Calcium Carbonate in Squeeze Process in White Tiger Oilfield. SPE J. 2004, 87467. [Google Scholar] [CrossRef]
- Da, S.; Kpsor, E.; Soletti, J.I.; De, C.E. Coalescence process to treat produced water: An updated overview and environmental outlook. Environ. Sci. Pollut. Res. Int. 2019, 26, 28668–28688. [Google Scholar]
- Amiri, M.; Moghadasi, J.; Jamialahmadi, M.; Shahri, M.P. Prediction of the Amount of Calcium Carbonate Scale Formation in Iranian Oilfields at Different Pressures. Energy Sources 2013, 35, 1197–1209. [Google Scholar] [CrossRef]
- Kapolos, J.; Koutsoukos, P.G. Formation of Calcium Phosphates in Aqueous Solutions in the Presence of Carbonate Ions. Langmuir 1999, 15, 6557–6562. [Google Scholar] [CrossRef]
- Charles, C.; David, C.; Arne, J. Measuring residence time distributions of wood chips in a screw conveyor reactor. Fuel Process. Technol. 2018, 178, 271–282. [Google Scholar]
- Koutsoukos, P.G.; Kapetanaki, E. Mixed calcium carbonate and calcium sulfate scale. In Proceedings of the corrosion 2016, Vancouver, BC, Canada, 6–10 March 2016. [Google Scholar]
- Mitchell, R.W.; Bowyer, P.M. Water injection methods. In Proceedings of the International Petroleum Exhibition and Technical Symposium of the Society of Ptoleum Engineers, Beijing, China, 17–23 March 1982. [Google Scholar]
- Sudmalis, M.; Sheikholeslami, R. Coprecipitation of CaCO3 and CaSO4. Can. J. Chem. Eng. 2010, 78, 21–31. [Google Scholar] [CrossRef]
- Klepetsanis, P.G.; Dalas, E.; Koutsoukos, P.G. Role of Temperature in the Spontaneous Precipitation of Calcium Sulfate Dihydrate. Langmuir 1999, 15, 1534–1540. [Google Scholar] [CrossRef]
- Lin, P.; Huang, S. The Effect of Temperatures on Calcium Carbonate Scale Formation. Chem. Eng. Oil Gas 1999, 28, 128–129. [Google Scholar]
- Yu, H.; Sheikholeslami, R.; Doherty, W. Mechanisms, thermodynamics and kinetics of composite fouling of calcium oxalate and amorphous silica in sugar mill evaporators—A preliminary study. Chem. Eng. Sci. 2002, 57, 1969–1978. [Google Scholar] [CrossRef]
- Helal, A.A.; Soames, A.D.; Gubner, R.; Iglauer, S.; Barifcani, A. Evaluating chemical-scale-inhibitor performance in external magnetic fields using a dynamic scale loop. J. Pet. Sci. Eng. 2019, 179, 1063–1077. [Google Scholar] [CrossRef]
- Davis, R.V.; Carter, P.W.; Kamrath, M.A.; Johnson, D.A.; Reed, P.E. The Use of Modern Methods in the Development of Calcium Carbonate Inhibitors for Cooling Water Systems; Springer: New York, NY, USA, 1995; pp. 33–46. [Google Scholar]
- Oddo, J.E.; Tomson, M.B. Why Scale Forms and How to Predict It. SPE Prod. Facil. 1994, 9, 47–54. [Google Scholar] [CrossRef]
- Alroomi, Y.M.; Hussain, K.F. Potential kinetic model for scaling and scale inhibition mechanism. Desalination 2016, 393, 186–195. [Google Scholar] [CrossRef]
- Al-Hamzah, A.A.; Wallace, A.D.; East, C.P.; Doherty, W.; Smith, E.; Fellows, C.M. Inhibition by Poly (acrylic acid) and Morphological Changes in Calcium Carbonate and Calcium Carbonate/Calcium Sulfate Crystallization on Silica Fibers. Ind. Eng. Chem. Res. 2014, 53, 8793–8803. [Google Scholar] [CrossRef]
- Elkholy, A.E.; Heakal, E.T.; Rashad, A.M.; Zakaria, K. Monte Carlo simulation for guar and xanthan gums as green scale inhibitors. J. Pet. Sci. Eng. 2018, 166, 263–273. [Google Scholar] [CrossRef]
- Senthilmurugan, B.; Ghosh, B.; Sanker, S. High performance maleic acid based oil well scale inhibitors—Development and comparative evaluation. J. Ind. Eng. Chem. 2011, 17, 415–420. [Google Scholar] [CrossRef]
- Sheikholeslami, R. Mixed salts—Scaling limits and propensity. Desalination 2003, 154, 117–127. [Google Scholar] [CrossRef]
- Natsi, P.D.; Rokidi, S.; Koutsoukos, P.G. Precipitation of calcium carbonate in aqueous solutions in presence of ethylene glycol and dodecane. Agric. Food Sci. J. Ghana 2010, 17. Available online: https://meetingorganizer.copernicus.org/EGU2015/EGU2015-9631.pdf (accessed on 20 November 2021).
- Cosmo, R.P.; Pereira, F.; Ribeiro, D.; Barros, W.Q.; Martins, A.L. Estimating CO2 degassing effect on CaCO3 precipitation under oil well conditions. J. Pet. Sci. Eng. 2019, 181, 106207. [Google Scholar] [CrossRef]
- Ghasemian, J.; Riahi, S.; Ayatollahi, S.; Mokhtari, R. Effect of salinity and ion type on formation damage due to inorganic scale deposition and introducing optimum salinity. J. Pet. Sci. Eng. 2019, 177, 270–281. [Google Scholar] [CrossRef]
- Sangwal, K. Induction period for crystallization. In Nucleation and Crystal Growth; John Wiley & Sons, Inc.: Hoboken, NY, USA, 2018; pp. 325–359. [Google Scholar]
- Hnel, O.S.; Mullin, J.W. Interpretation of Crystallization Induction Peroids. J. Colloid Interface Sci. 1988, 123, 43–50. [Google Scholar]
- Tadier, S.; Rokidi, S.; Rey, C.; Combes, C.; Koutsoukos, P.G. Crystal growth of aragonite in the presence of phosphate. J. Cryst. Growth 2016, 458, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Bursaux, R.; Peltier, S.; Nguyen, M.; Romero, C.; Morel, D. Single well tracer test results in a high temperature, high salinity offshore carbonate reservoir for chemical eor pilot evaluation. In Proceedings of the SPE Improved Oil Recovery Conference, Tulsa, OK, USA, 11–13 April 2016. [Google Scholar]
- Nancollas, H.G.; Reddy, M.M. The crystallization of calcium carbonate. II. Calcite growth mechanism. J. Colloid Interface Sci. 1971, 37, 824–830. [Google Scholar] [CrossRef]
- Khormali, A.; Petrakov, D.G.; Lamidi, A.L.B.; Rastegar, R. Prevention of calcium carbonate precipitation during water injection into high-pressure high-temperature wells. In Proceedings of the SPE European Formation Damage Conference and Exhibition, Budapest, Hungary, 3–5 June 2015. [Google Scholar]
- Sheikholeslami, R.; Ng, M. Calcium Sulfate Precipitation in the Presence of Nondominant Calcium Carbonate: Thermodynamics and Kinetics. Ind. Eng. Chem. Res. 2001, 40, 3570–3578. [Google Scholar] [CrossRef]
- Gopi, S.P.; Subramanian, V.; Palanisamy, K. Synergistic Effect of EDTA and HEDP on the Crystal Growth, Polymorphism, and Morphology of CaCO3. Ind. Eng. Chem. Res. 2015, 54, 3618–3625. [Google Scholar] [CrossRef]
- Gadri, F.A.E. Kinetics of the precipitation of calcium sulfate dihydrate in a desalination unit. Desalination 2003, 158, 9–16. [Google Scholar]
- Nancollas, G.H.W.W. The Kinetics and Mechanism of Formation of Calcium Sulfate Scale Minerals. Corrosion 1979, 35, 304–308. [Google Scholar] [CrossRef]
- Amiri, M.; Moghadasi, J.; Jamialahmadi, M. Prediction of Iron Carbonate Scale Formation in Iranian Oilfields at Different Mixing Ratio of Injection Water with Formation Water. Energy Sources 2013, 35, 1256–1265. [Google Scholar] [CrossRef]
- Sheikholeslami, R.; Ong, H. Kinetics and thermodynamics of calcium carbonate and calcium sulfate at salinities up to 1.5 M. Desalination 2003, 157, 217–234. [Google Scholar] [CrossRef]
- Melliti, E.; Touati, K.; Bruggen, B.; Elfil, H. Effect of Fe2+ ions on Gypsum Precipitation During Bulk Crystallization of Reverse Osmosis Concentrates. Chemosphere 2020, 263, 127866. [Google Scholar] [CrossRef] [PubMed]
- Sheikholeslami, T. Thermodynamics and kinetics for mixed calcium carbonate and calcium sulfate precipitation. Chem. Eng. Sci. 2001, 56, 5391–5400. [Google Scholar]
- Liu, S.T.; Nancollas, G.H. The crystallization of magnesium hydroxide. Desalination 1973, 12, 75–84. [Google Scholar]
- Olajire, A.A. A review of oilfield scale management technology for oil and gas production. J. Pet. Sci. Eng. 2015, 135, 723–737. [Google Scholar] [CrossRef]
- Shahzad, K.M.; Ibnelwaleed, H.; Mohamed, M.; Sultan, A.S.; Saad, M. Oilfield scale formation and chemical removal: A review. J. Pet. Sci. Eng. 2018, 171, 127–139. [Google Scholar]
- Touati, K.; Alia, E.; Zendah, H.; Elfil, H.; Hannachi, A. Sand filters scaling by calcium carbonate precipitation during groundwater reverse osmosis desalination. Desalination 2018, 430, 24–32. [Google Scholar] [CrossRef]
- Baumgartner, J.; Dey, A.; Bomans, P.; Coadou, C.; Fratzl, P.; Sommerdijk, N.; Faivre, D. Nucleation and growth of magnetite from solution. Nat. Mater. 2013, 12, 310–314. [Google Scholar] [CrossRef]
- Habraken, W.J.E.M.; Tao, J.; Brylka, L.J.; Friedrich, H.; Bertinetti, L.; Schenk, A.S.; Verch, A.; Dmitrovic, V.; Bomans, P.H.H.; Frederik, P.M.; et al. Ion-association complexes unite classical and non-classical theories for the biomimetic nucleation of calcium phosphate. Nat. Commun. 2013, 4, 1507. [Google Scholar] [CrossRef] [Green Version]
- Radha, A.V.; Forbes, T.Z.; Killian, C.E.; Gilbert, P.; Navrotsky, A. Crystallization energetics of amorphous calcium carbonate. Proc. Natl. Acad. Sci. USA 2010, 107, 16438–16443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nancollas, G.H.; Reddy, M.M. Crystal growth kinetics of minerals encountered in water treatment processes. In Aqueous-Environmental Chemistry of Metals; Rubin, A.J., Ed.; Ann Arbor Science Publishers, Inc.: Ann Arbor, MI, USA, 1973. [Google Scholar]
- Nancollas, G.H.; Reddy, M.M. The crystallization of calcium carbonate: I. Isotopic exchange and kinetics. J. Colloid Interface Sci. 1971, 37, 166–172. [Google Scholar]
- Chaturvedi, E.; Patidar, K.; Srungavarapu, M.; Laik, S.; Mandal, A. Thermodynamics and kinetics of methane hydrate formation and dissociation in presence of calcium carbonate. Adv. Powder Technol. 2018, 29, S0921883118300347. [Google Scholar] [CrossRef]
- Bramson, D.; Hasson, D.; Semiat, R. The roles of gas bubbling, wall crystallization and particulate deposition in CaSO4 scale formation. Desalination 1995, 100, 105–113. [Google Scholar] [CrossRef]
- Bots, P.; Benning, L.G.; Rodriguez-Blanco, J.D.; Roncal-Herrero, T.; Shaw, S. Mechanistic Insights into the Crystallization of Amorphous Calcium Carbonate (ACC). Cryst. Growth Des. 2012, 12, 3806–3814. [Google Scholar] [CrossRef]
- Yoreo, J.D. Crystal nucleation: More than one pathway. Nat. Mater. 2013, 12, 284–285. [Google Scholar] [CrossRef]
- Augustin, W.; Bohnet, M. Influence of the ratio of free hydrogen ions on crystallization fouling. Chem. Eng. Process. 1995, 34, 79–85. [Google Scholar] [CrossRef]
- Sun, R.; Willhammar, T.; Grape, E.S.; Strmme, M.; Cheung, O. Mesoscale Transformation of Amorphous Calcium Carbonate to Porous Vaterite Microparticles with Morphology Control. Cryst. Growth Des. 2019, 19, 5075–5087. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, Z.; Liu, J.; Kerisit, S.N.; Bowden, M.E.; Sushko, M.L. Direction-specific interaction forces underlying zinc oxide crystal growth by oriented attachment. Nat. Commun. 2017, 8, 835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Saivenkataraman, J.; Chen, W.; Kristin, A.; Gerbrand, P. Nucleation of metastable aragonite CaCO3 in seawater. Proc. Nat. Acad. Sci. USA 2015, 112, 3199–3204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nebel, H.; Neumann, M.; Mayer, C.; Epple, M. On the structure of Amorphous Calcium Carbonate—A detailed study by solid-state NMR spectroscopy. Inorg. Chem. 2008, 47, 7874–7879. [Google Scholar] [CrossRef]
- Plummer, L.N.; Parkhurst, D.L.; Wigley, T.M.L. Critical review of the kinetics of calcite dissolution and precipitation. In Chemical Modeling in Aqueous Systems; ACS Publications: Washington, DC, USA, 1979. [Google Scholar]
- Rodriguez-Blanco, J.D.; Shaw, S.; Benning, L.G. The Kinetics and Mechanisms of Amorphous Calcium Carbonate (ACC) Crystallization to Calcite, Via Vaterite. Nanoscale 2010, 3, 265–271. [Google Scholar] [CrossRef]
- Fernández-Díaz, L.; Fernandez-Gonzalez, M.D.L.A.; Prieto, M. The role of sulfate groups in controlling CaCO3 polymorphism. Geochim. Cosmochim. Acta 2010, 74, 6064–6076. [Google Scholar] [CrossRef] [Green Version]
- Nikos, S.; Petros, G.K. The transformation of vaterite to calcite: Effect of the conditions of the solutions in contact with the mineral phase. J. Cryst. Growth 1998, 191, 783–790. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Ca | Sulfate | Carbonate | Precipitates |
|---|---|---|---|---|
| 1 | 0.145 mol/L (100 mL) | 0.145 mol/L (100 mL) | – | gypsum |
| 2 | 0.145 mol/L (100 mL) | – | 0.29 mol/L (100 mL) | calcite |
| 3 | 0.145 mol/L (100 mL) | 0.145 mol/L (50 mL) | 0.29 mol/L (50 mL) | mixed scale |
| 4 | 0.145 mol/L (100 mL) | 0.24 mol/L (50 mL) | 0.1 mol/L (50 mL) | mixed scale |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, Y.; Yu, T.; Zhang, H.; Song, J.; Qu, C.; Li, J.; Yang, B. Co-Deposition Mechanisms of Calcium Sulfate and Calcium Carbonate Scale in Produced Water. Crystals 2021, 11, 1494. https://doi.org/10.3390/cryst11121494
Yan Y, Yu T, Zhang H, Song J, Qu C, Li J, Yang B. Co-Deposition Mechanisms of Calcium Sulfate and Calcium Carbonate Scale in Produced Water. Crystals. 2021; 11(12):1494. https://doi.org/10.3390/cryst11121494
Chicago/Turabian StyleYan, Yan, Tao Yu, Huan Zhang, Jiayu Song, Chengtun Qu, Jinling Li, and Bo Yang. 2021. "Co-Deposition Mechanisms of Calcium Sulfate and Calcium Carbonate Scale in Produced Water" Crystals 11, no. 12: 1494. https://doi.org/10.3390/cryst11121494