
Dependence of Crystallization Behavior of Interacting Telechelic Poly(butylene succinate) Oligomer on Molecular Weight


Abstract
:1. Introduction
2. Experimental
2.1. Materials
2.2. Synthesis of UPy-NCO
2.3. Synthesis of PBS-OH
2.4. Synthesis of PBS-UPy
2.5. Characterization
3. Results
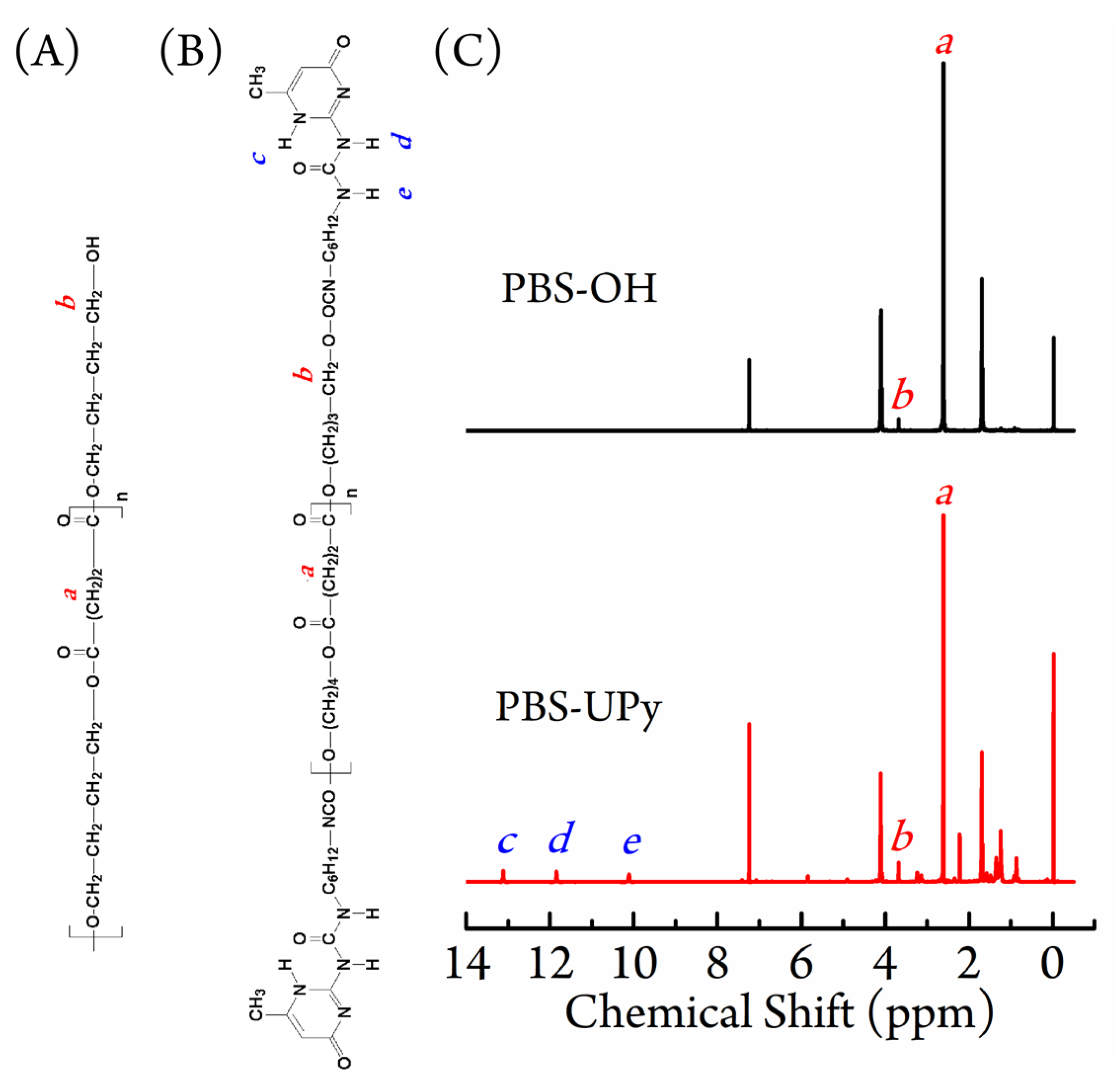
3.1. Chain Structure
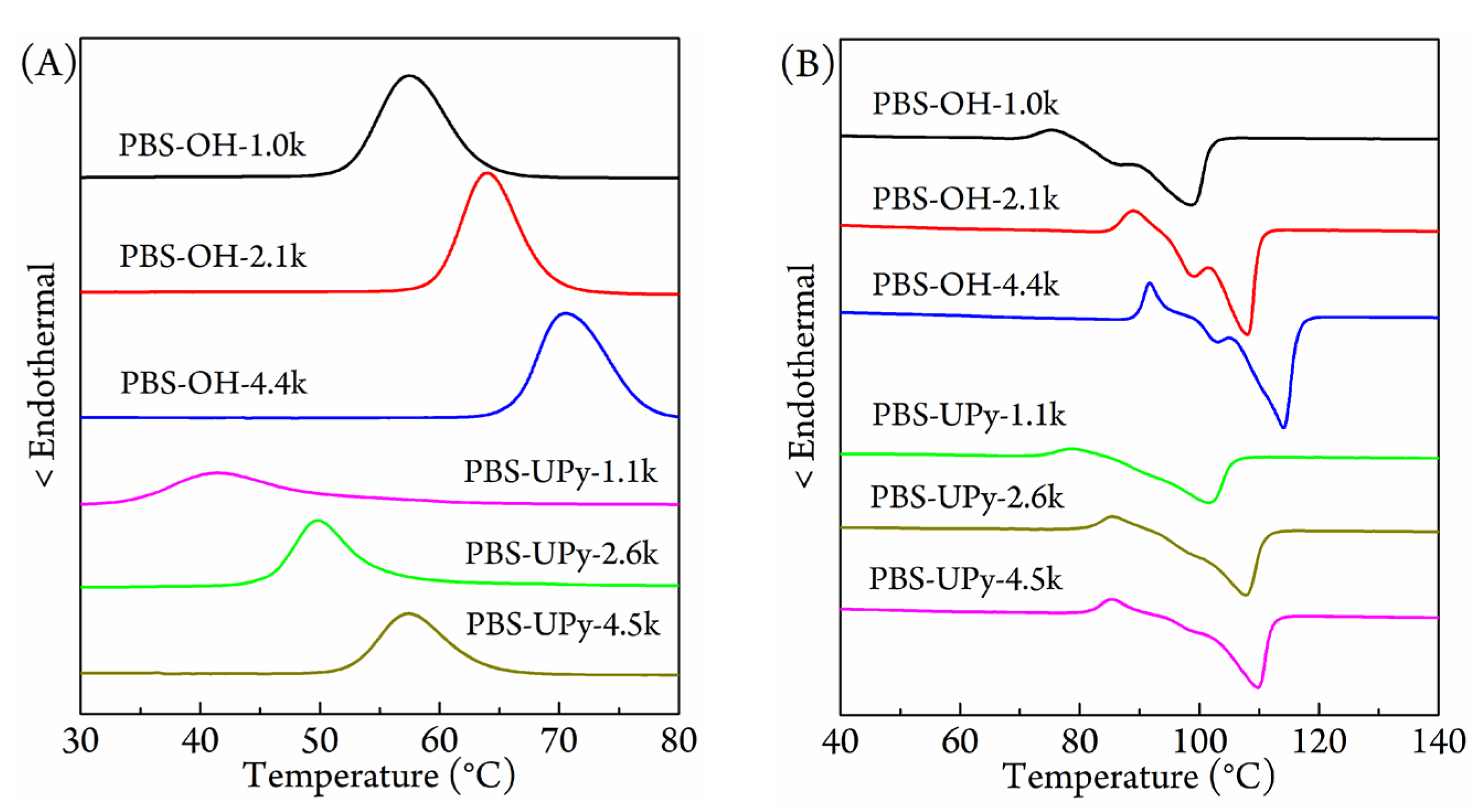
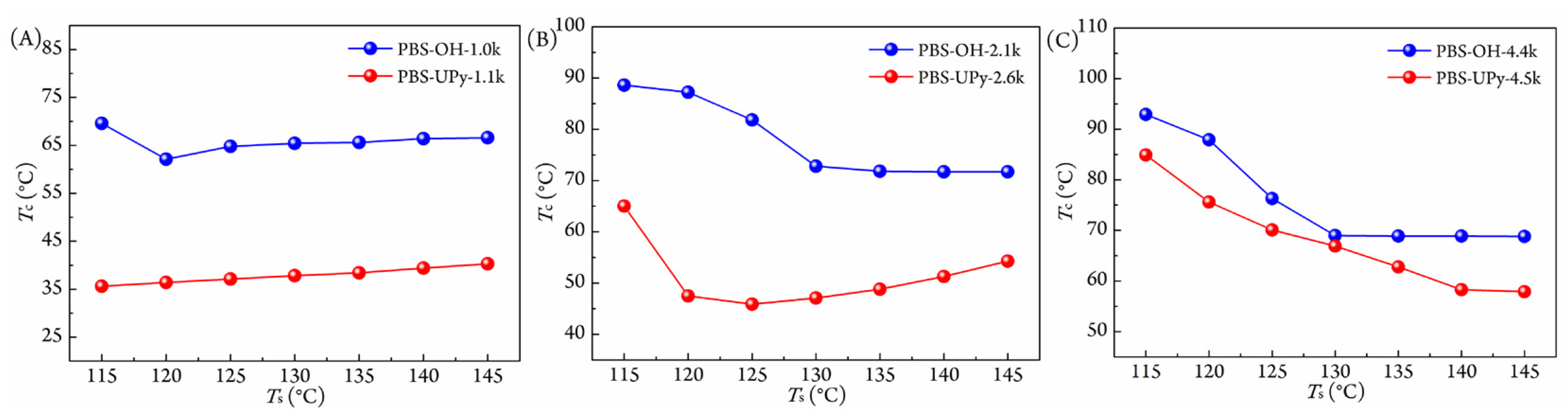
3.2. Non-Isothermal Crystallization
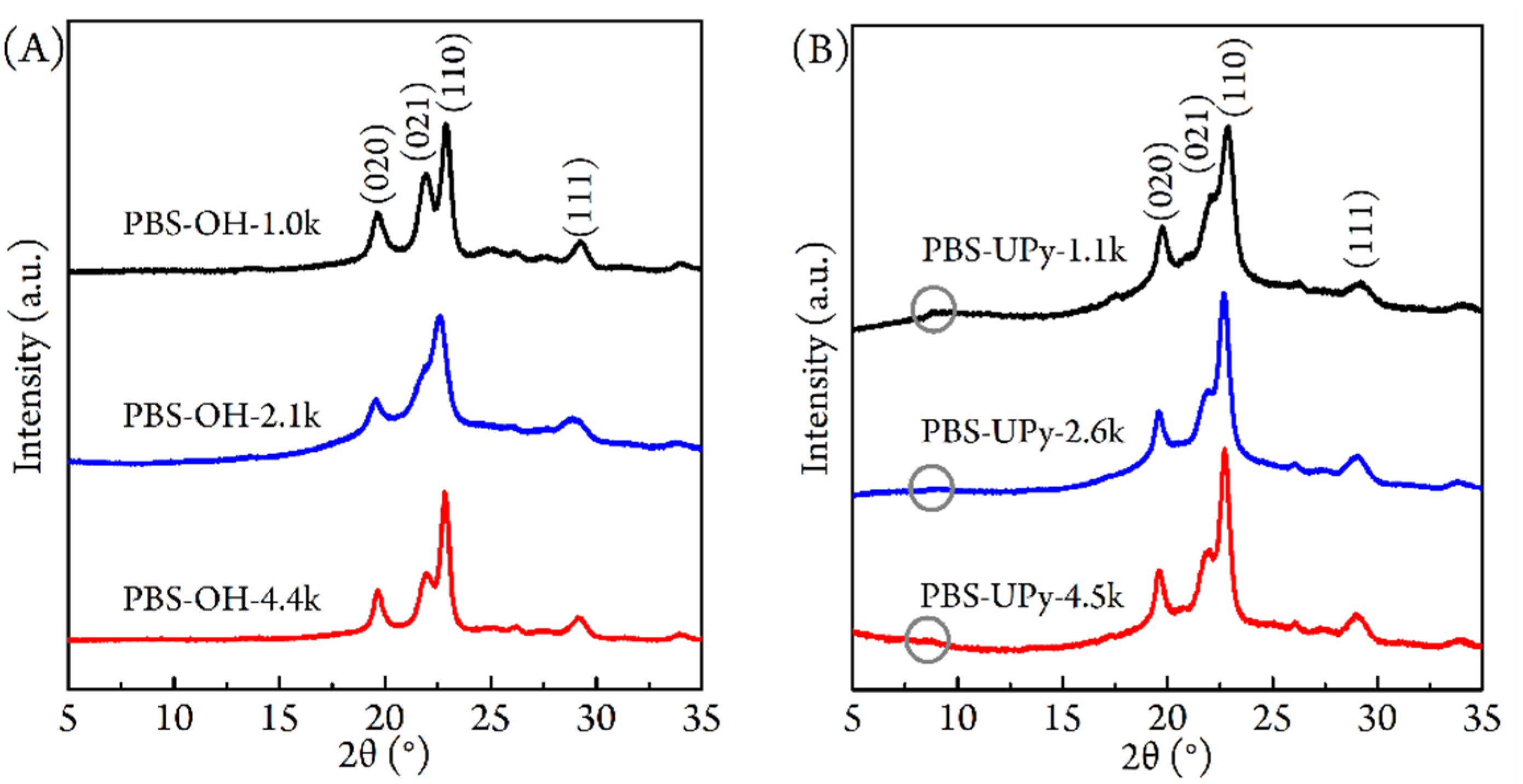
3.3. Crystalline Structure and Morphology
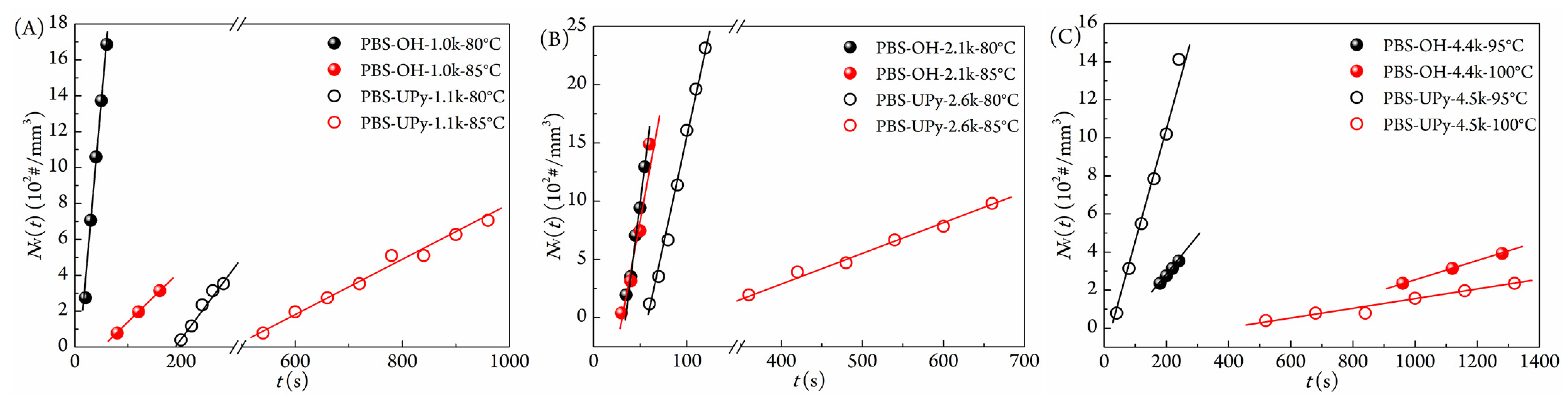
3.4. Nucleation Behavior and Melt Memory Effect
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahn, B.D.; Kim, S.H.; Kim, Y.H.; Yang, J.S. Synthesis and characterization of the biodegradable copolymers from succinic acid and adipic acid with 1,4-butanediol. J. Appl. Polym. Sci. 2001, 82, 2808–2826. [Google Scholar] [CrossRef]
- Miyata, T.; Masuko, T. Crystallization behaviour of poly(tetramethylene succinate). Polymer 1998, 39, 1399–1404. [Google Scholar] [CrossRef]
- Yoo, E.S.; Im, S.S. Melting behavior of poly(butylene succinate) during heating scan by DSC. J. Polym. Sci. Part B Polym. Phys. 1999, 37, 1357–1366. [Google Scholar] [CrossRef]
- Liu, W.G.; Zhang, X.C.; Li, H.Y.; Liu, Z. Effect of surface modification with 3-aminopropyltriethyloxy silane on mechanical and crystallization performances of ZnO/poly(butylene succinate) composites. Compos. Part B-Eng. 2012, 43, 2209–2216. [Google Scholar] [CrossRef]
- Pandey, J.K.; Reddy, K.R.; Kumar, A.P.; Singh, R.P. An overview on the degradability of polymer nanocomposites. Polym. Degrad. Stab. 2005, 88, 234–250. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, M.; Song, J.; Miao, Z.C. Effects of quaternization on properties of biodegradable Poly(butylene succinate) with low crystallinity. Mater. Res. Express 2019, 6, 045304. [Google Scholar] [CrossRef]
- Li, Y.; Huang, G.Y.; Chen, C.; Wei, X.W.; Dong, X.; Zhao, W.; Ye, H.M. Poly(butylene succinate-co-butylene acetylenedicarboxylate): Copolyester with Novel Nucleation Behavior. Polymers 2021, 13, 365. [Google Scholar] [CrossRef] [PubMed]
- Arandia, I.; Zaldua, N.; Maiz, J.; Pérez-Camargo, R.A.; Mugica, A.; Zubitur, M.; Mincheva, R.; Dubois, P.; Müller, A.J. Tailoring the isothermal crystallization kinetics of isodimorphic poly(butylene succinate-ran-butylene azelate) random copolymers by changing composition. Polymer 2019, 183, 121863. [Google Scholar] [CrossRef]
- Chae, H.G.; Park, S.H.; Kim, B.C.; Kim, D.K. Effect of methyl substitution of the ethylene unit on the physical properties of poly(butylene succinate). J. Polym. Sci. Part B Polym. Phys. 2004, 42, 1759–1766. [Google Scholar] [CrossRef]
- Chae, D.W.; Kim, B.C.; Kim, D.K. Effects of shearing and comonomer content on the crystallization behavior of poly(butylene succinate-co-butylene 2-ethyl-2-methyl succinate). Polym. Int. 2004, 53, 1266–1273. [Google Scholar] [CrossRef]
- Ye, H.M.; Wang, R.D.; Liu, J.; Xu, J.; Guo, B.H. Isomorphism in Poly(butylene succinate-co-butylene fumarate) and Its Application as Polymeric Nucleating Agent for Poly(butylene succinate). Macromolecules 2012, 45, 5667–5675. [Google Scholar] [CrossRef]
- Yang, B.; Ni, H.; Huang, J.; Luo, Y. Effects of Poly(vinyl butyral) as a Macromolecular Nucleating Agent on the Nonisothermal Crystallization and Mechanical Properties of Biodegradable Poly(butylene succinate). Macromolecules 2014, 47, 284–296. [Google Scholar] [CrossRef]
- Tang, Y.R.; Lin, D.W.; Gao, Y.; Xu, J.; Guo, B.H. Prominent Nucleating Effect of Finely Dispersed Hydroxyl-Functional Hexagonal Boron Nitride on Biodegradable Poly(butylene succinate). Ind. Eng. Chem. Res. 2014, 53, 4689–4696. [Google Scholar] [CrossRef]
- Yarici, T.; Kodal, M.; Ozkoc, G. Non-isothermal crystallization kinetics of Poly(butylene succinate) (PBS) nanocomposites with different modified carbon nanotubes. Polymer 2018, 146, 361–377. [Google Scholar] [CrossRef]
- Hall, K.W.; Percec, S.; Shinoda, W.; Klein, M.W. Chain-End Modification: A Starting Point for Controlling Polymer Crystal Nucleation. Macromolecules 2021, 54, 1599–1610. [Google Scholar] [CrossRef]
- Bao, J.N.; Fan, H.B.; Xue, X.J.; Xie, Q.; Pan, P.J. Temperature-Dependent Crystalline Structure and Phase Transition of Poly(butylene adipate) End-Functionalized by Multiple Hydrogen-Bonding Groups. Phys. Chem. Chem. Phys. 2018, 20, 26479–26488. [Google Scholar] [CrossRef]
- Li, X.; Xu, W.Q.; Yuan, W.H.; Liu, K.K.; Zhou, J.; Shan, G.R.; Bao, Y.Z.; Pan, P.J. Separate crystallization and melting of polymer blocks and hydrogen bonding units in double-crystalline supramolecular polymers. Polymer 2021, 222, 123670. [Google Scholar] [CrossRef]
- Bao, J.N.; Chang, X.H.; Xie, Q.; Yu, C.T.; Shan, G.R.; Bao, Y.Z.; Pan, P.J. Preferential Formation of β-Form Crystals and Temperature-Dependent Polymorphic Structure in Supramolecular Poly(l-lactic Acid) Bonded by Multiple Hydrogen Bonds. Macromolecules 2017, 50, 8619–8630. [Google Scholar] [CrossRef]
- Bao, J.N.; Chang, R.X.; Shan, G.R.; Bao, Y.Z.; Pan, P.J. Promoted Stereocomplex Crystallization in Supramolecular Stereoblock Copolymers of Enantiomeric Poly(lactic acid)s. Cryst. Growth Des. 2016, 16, 1502–1511. [Google Scholar] [CrossRef]
- Li, X.; Ni, L.L.; Sun, C.X.; Xu, W.Q.; Zheng, Y.; Shan, G.R.; Bao, Y.Z.; Pan, P.J. Nucleobase-monofunctionalized supramolecular poly(l-lactide): Controlled synthesis, competitive crystallization, and structural organization. Polym. Chem. 2021, 12, 3461. [Google Scholar] [CrossRef]
- Xu, S.S.; Li, X.; Sun, C.X.; Ni, L.L.; Xu, W.Q.; Yuan, W.H.; Zheng, Y.; Yu, C.T.; Pan, P.J. Controllable crystallization and lamellar organization in nucleobase-functionalized supramolecular poly(lactic acid)s: Role of poly(lactic acid) stereostructure. Polymer 2021, 232, 124148. [Google Scholar] [CrossRef]
- Yu, M.M.; Yang, W.J.; Niu, D.Y.; Cai, X.X.; Weng, Y.X.; Dong, W.F.; Chen, M.Q.; Xu, P.W.; Wang, Y.; Chu, H.; et al. Enhancing the crystallization performance of poly(l-lactide) by intramolecular hybridizing with tunable self-assembly-type oxalamide segments. Chin. J. Polym. Sci. 2021, 39, 122–132. [Google Scholar] [CrossRef]
- Folmer, B.J.B.; Sijbesma, R.P.; Versteegen, R.M.; van der Rijt, J.A.J.; Meijer, E.W. Supramolecular Polymer Materials: Chain Extension of Telechelic Polymers Using a Reactive Hydrogen-Bonding Synthon. Adv. Mater. 2000, 12, 874–878. [Google Scholar] [CrossRef]
- Zheng, L.C.; Li, C.C.; Wang, Z.D.; Wang, J.; Xiao, Y.N.; Zhang, D.; Guan, G.H. Novel Biodegradable and Double Crystalline Multiblock Copolymers Comprising of Poly(butylene succinate) and Poly(ε-caprolactone): Synthesis, Characterization, and Properties. Ind. Eng. Chem. Res. 2012, 51, 7264–7272. [Google Scholar] [CrossRef]
- Dankers, P.Y.W.; Zhang, Z.; Wisse, E.; Grijpma, D.W.; Sijbesma, R.P.; Feijen, J.; Meijer, E.W. Oligo(trimethylene carbonate)-based supramolecular biomaterials. Macromolecules 2006, 39, 8763–8771. [Google Scholar] [CrossRef]
- Liu, B.; Tang, Z.; Wang, Z.; Zhang, L.; Guo, B. Integrating transient and sacrificial bonds into biobased elastomers toward mechanical property enhancement and macroscopically responsive property. Polymer 2019, 184, 121914. [Google Scholar] [CrossRef]
- Liu, G.C.; Zhang, W.Q.; Zhou, S.L.; Wang, X.L. Improving crystallization and processability of PBS via slight cross-linking. RSC Adv. 2016, 6, 68942. [Google Scholar] [CrossRef]
- Gan, Z.; Abe, H.; DOi, Y. Crystallization, Melting, and Enzymatic Degradation of Biodegradable Poly(butylene succinate-co-14 mol% ethylene succinate) Copolyester. Biomacromolecules 2001, 2, 313–321. [Google Scholar] [CrossRef]
- Zhang, R.; Zhuravlev, E.; Schmelzer, J.; Androsch, R.; Schick, C. Steady-State Crystal Nucleation Rate of Polyamide 66 by Combining Atomic Force Microscopy and Fast-Scanning Chip Calorimetry. Macromolecules 2020, 53, 5560–5571. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mn (g/mol) | Sample | Mn (g/mol) |
|---|---|---|---|
| PBS-OH-1.0k | 1.0 × 103 | PBS-UPy-1.1k | 1.1 × 103 |
| PBS-OH-2.1k | 2.1 × 103 | PBS-UPy-2.6k | 2.6 × 103 |
| PBS-OH-4.4k | 4.4 × 103 | PBS-UPy-4.5k | 4.5 × 103 |
| Sample | Tc (°C) | ∆Hc (J/g) | Tm (°C) | ∆Hm (J/g) |
|---|---|---|---|---|
| PBS-OH-1.0k | 57.5 | 70.4 | 98.7 | 70.8 |
| PBS-OH-2.1k | 63.9 | 71.2 | 108.0 | 73.9 |
| PBS-OH-4.4k | 70.5 | 73.2 | 114.2 | 74.3 |
| PBS-UPy-1.1k | 41.6 | 38.6 | 101.6 | 47.1 |
| PBS-UPy-2.6k | 49.8 | 46.7 | 107.6 | 53.7 |
| PBS-UPy-4.5k | 57.5 | 43.3 | 109.8 | 47.5 |
| Sample | Tc (°C) | Jst (#/(mm3·s)) | tind (s) |
|---|---|---|---|
| PBS-OH-1.0k | 80 | 34.9 | 11 |
| 85 | 2.94 | 53 | |
| PBS-UPy-1.1k | 80 | 4.12 | 189 |
| 85 | 1.47 | 473 | |
| PBS-OH-2.1k | 80 | 55.7 | 32 |
| 85 | 47.8 | 31 | |
| PBS-UPy-2.6k | 80 | 40.4 | 62 |
| 85 | 2.52 | 279 | |
| PBS-OH-4.4k | 95 | 2.75 | 106 |
| 100 | 0.49 | 480 | |
| PBS-UPy-4.5k | 95 | 6.44 | 32 |
| 100 | 0.25 | 402 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Zhang, X.-W.; Ye, H.-M. Dependence of Crystallization Behavior of Interacting Telechelic Poly(butylene succinate) Oligomer on Molecular Weight. Crystals 2021, 11, 1530. https://doi.org/10.3390/cryst11121530
Chen C, Zhang X-W, Ye H-M. Dependence of Crystallization Behavior of Interacting Telechelic Poly(butylene succinate) Oligomer on Molecular Weight. Crystals. 2021; 11(12):1530. https://doi.org/10.3390/cryst11121530
Chicago/Turabian StyleChen, Cong, Xue-Wen Zhang, and Hai-Mu Ye. 2021. "Dependence of Crystallization Behavior of Interacting Telechelic Poly(butylene succinate) Oligomer on Molecular Weight" Crystals 11, no. 12: 1530. https://doi.org/10.3390/cryst11121530