
Effects of Cr Doping and Water Content on the Crystal Structure Transitions of Ba2In2O5


 , , ,
, , , 

Abstract
:
1. Introduction
2. Materials, Methods, and Equipment
2.1. Synthesis
2.2. Thermal Analysis
2.3. Powder X-ray Diffraction
2.4. Powder Neutron Diffraction
2.5. Optical Spectroscopy
2.6. Solid-State Nuclear Resonance Spectroscopy
2.7. Software
3. Results
3.1. Thermal Analysis
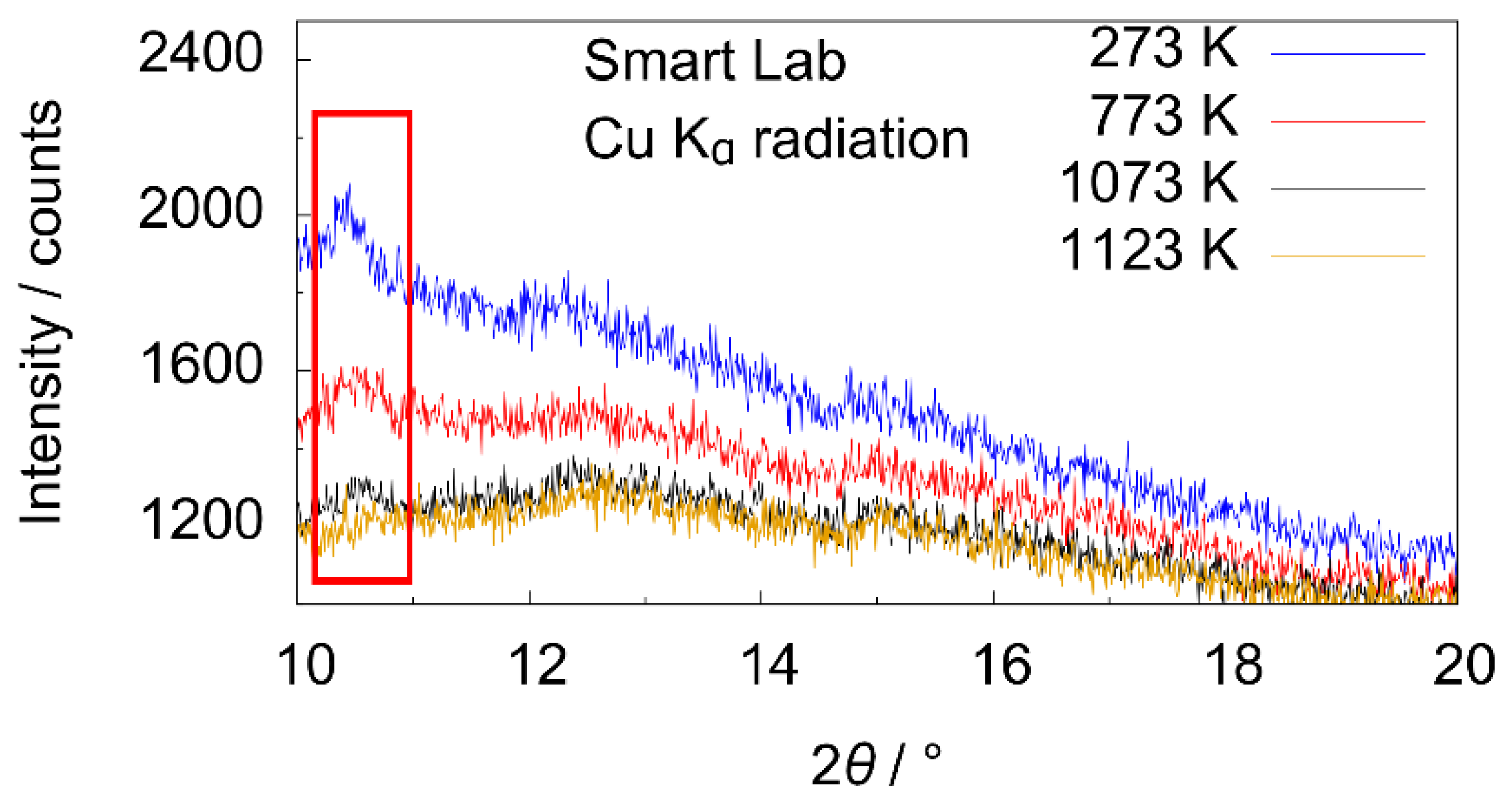
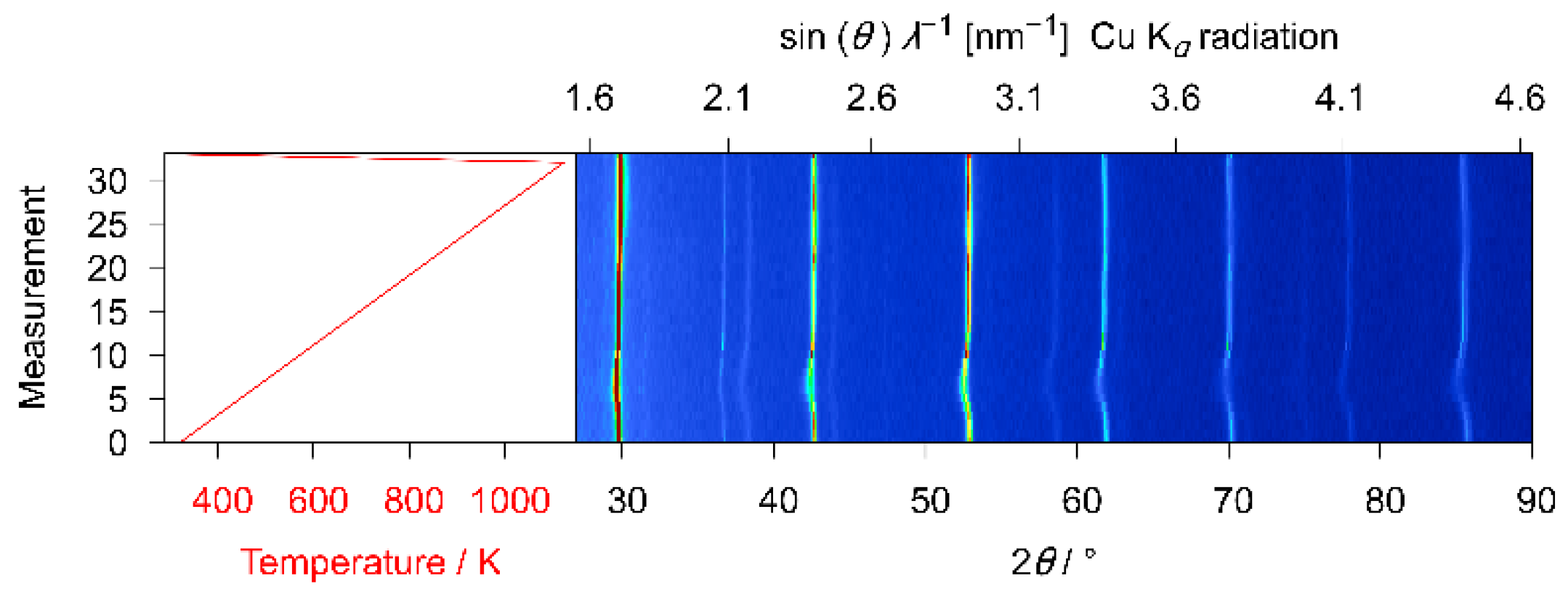
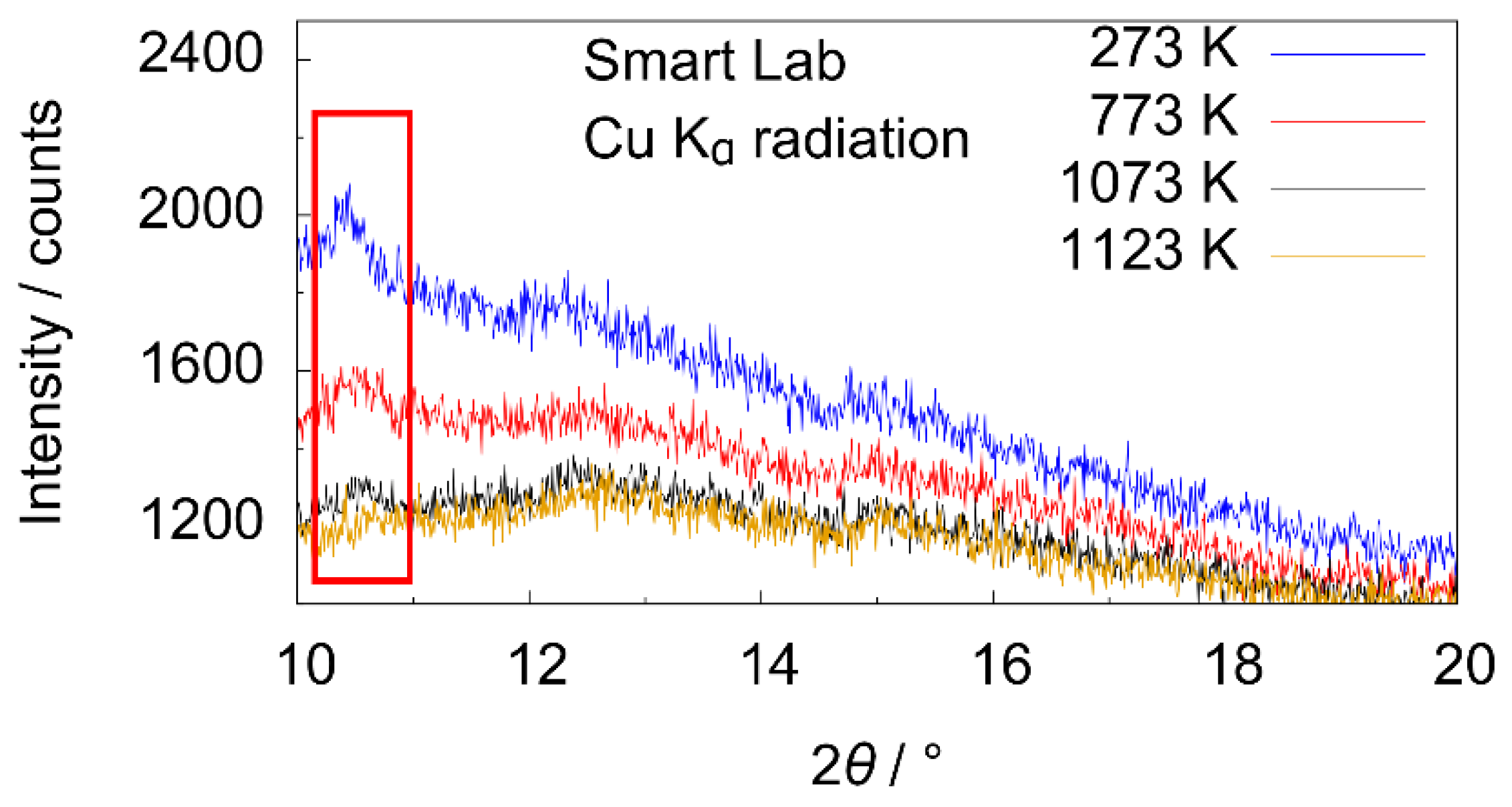
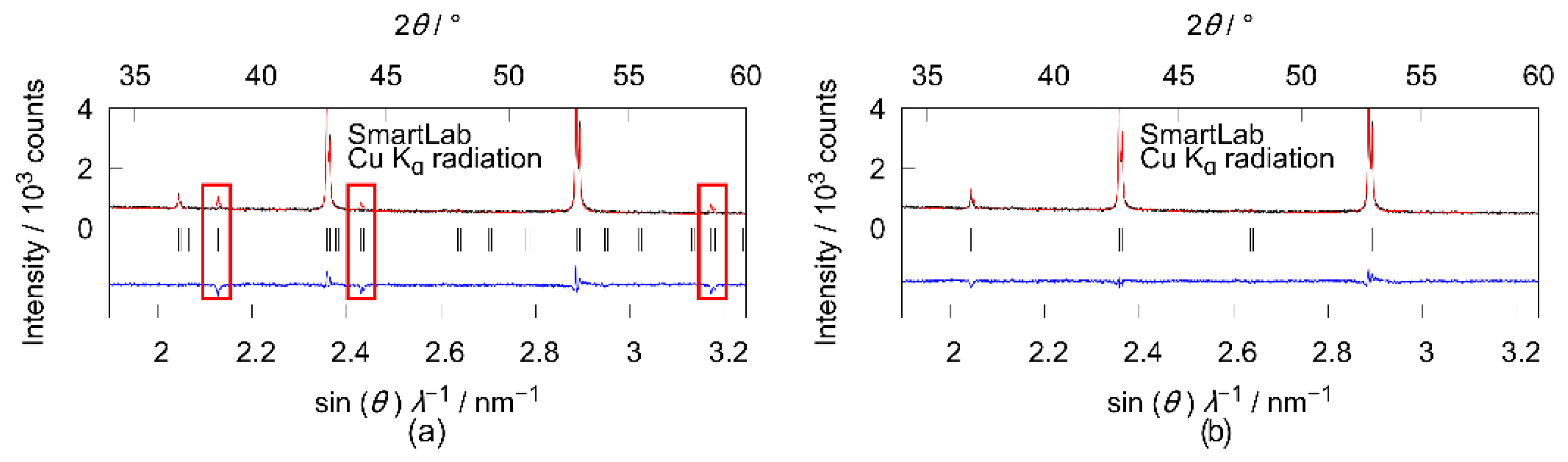
3.2. X-ray Diffraction
3.3. Neutron Diffraction Experiments and Nuclear Magnetic Resonance Spectroscopy
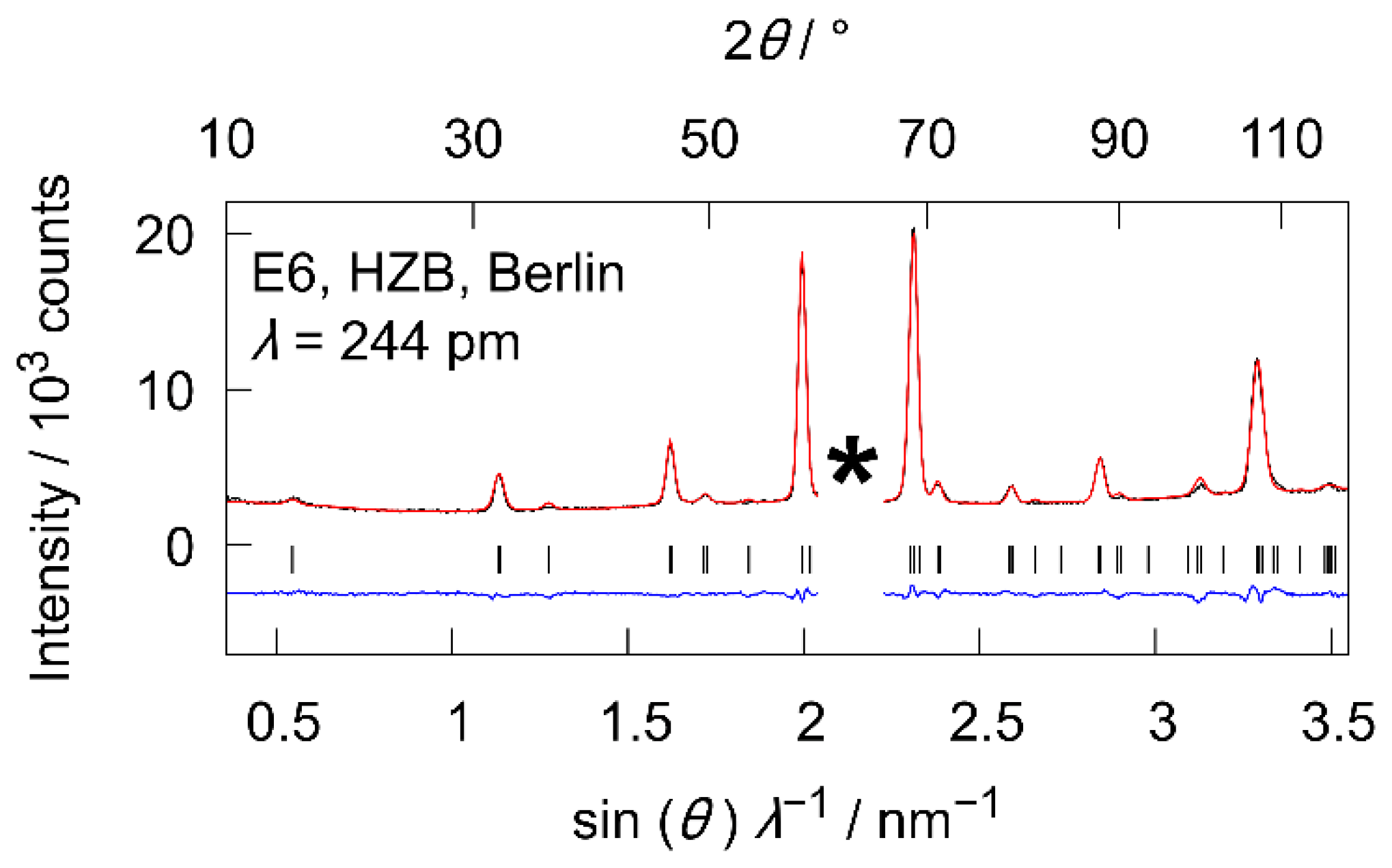
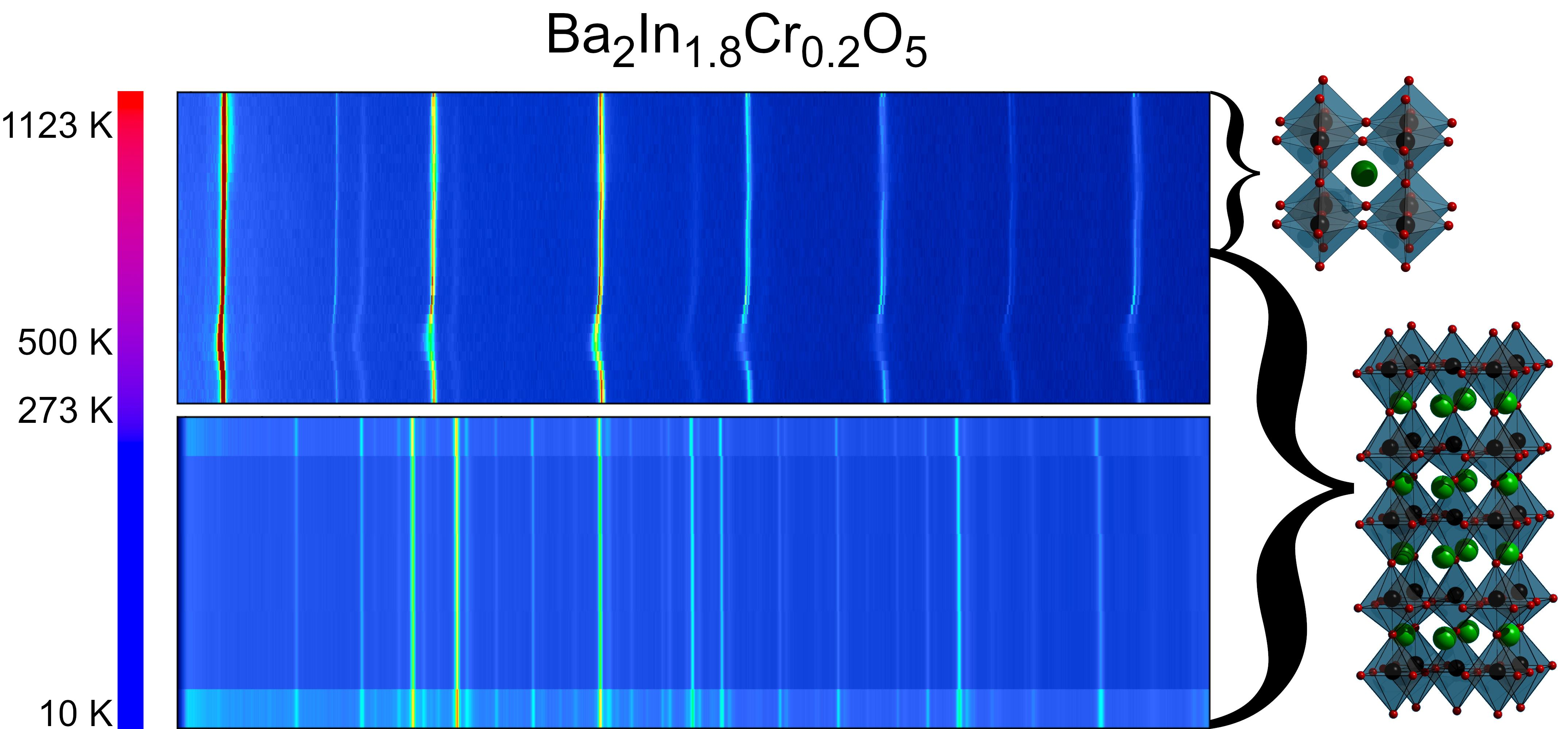
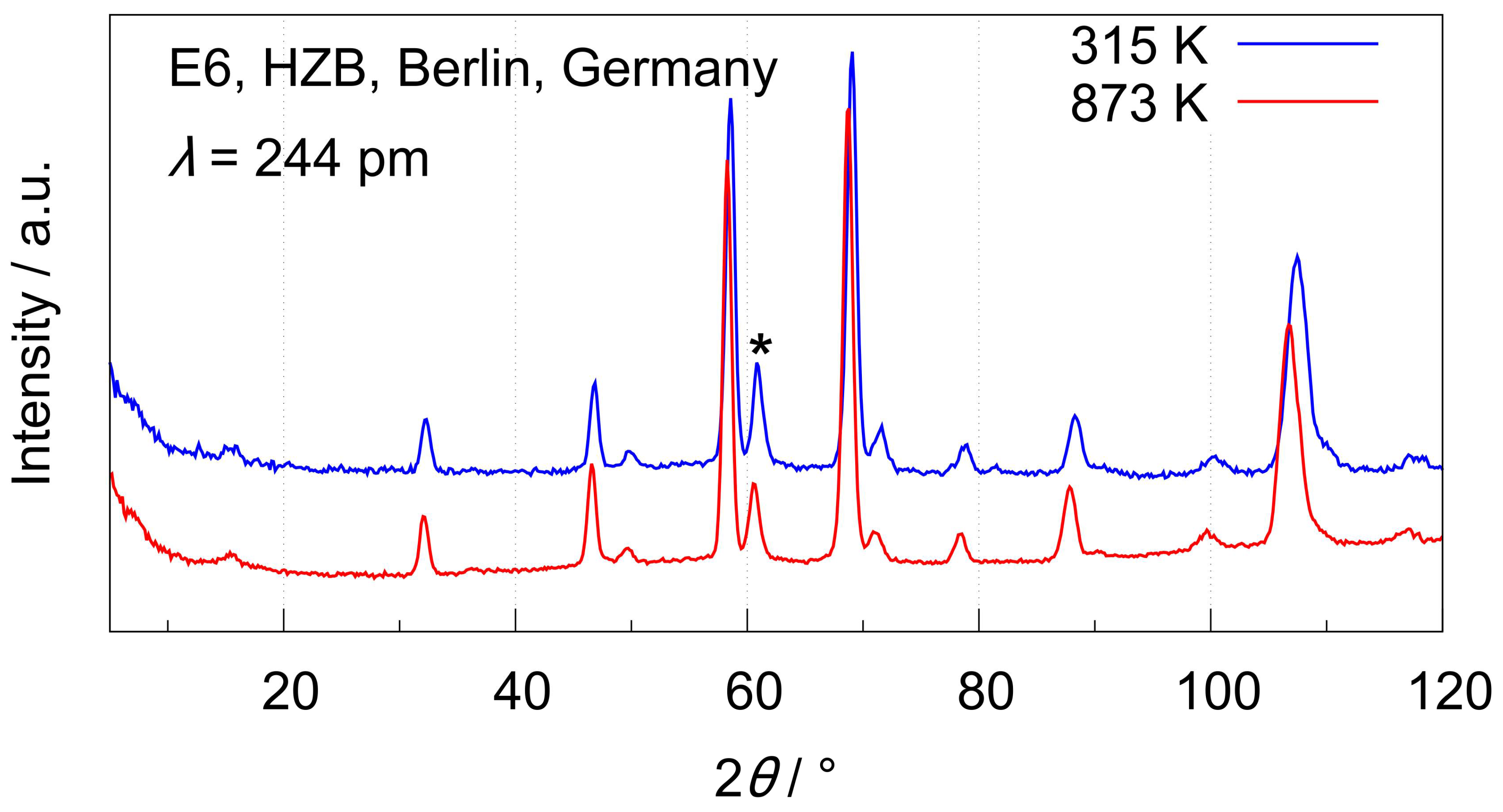
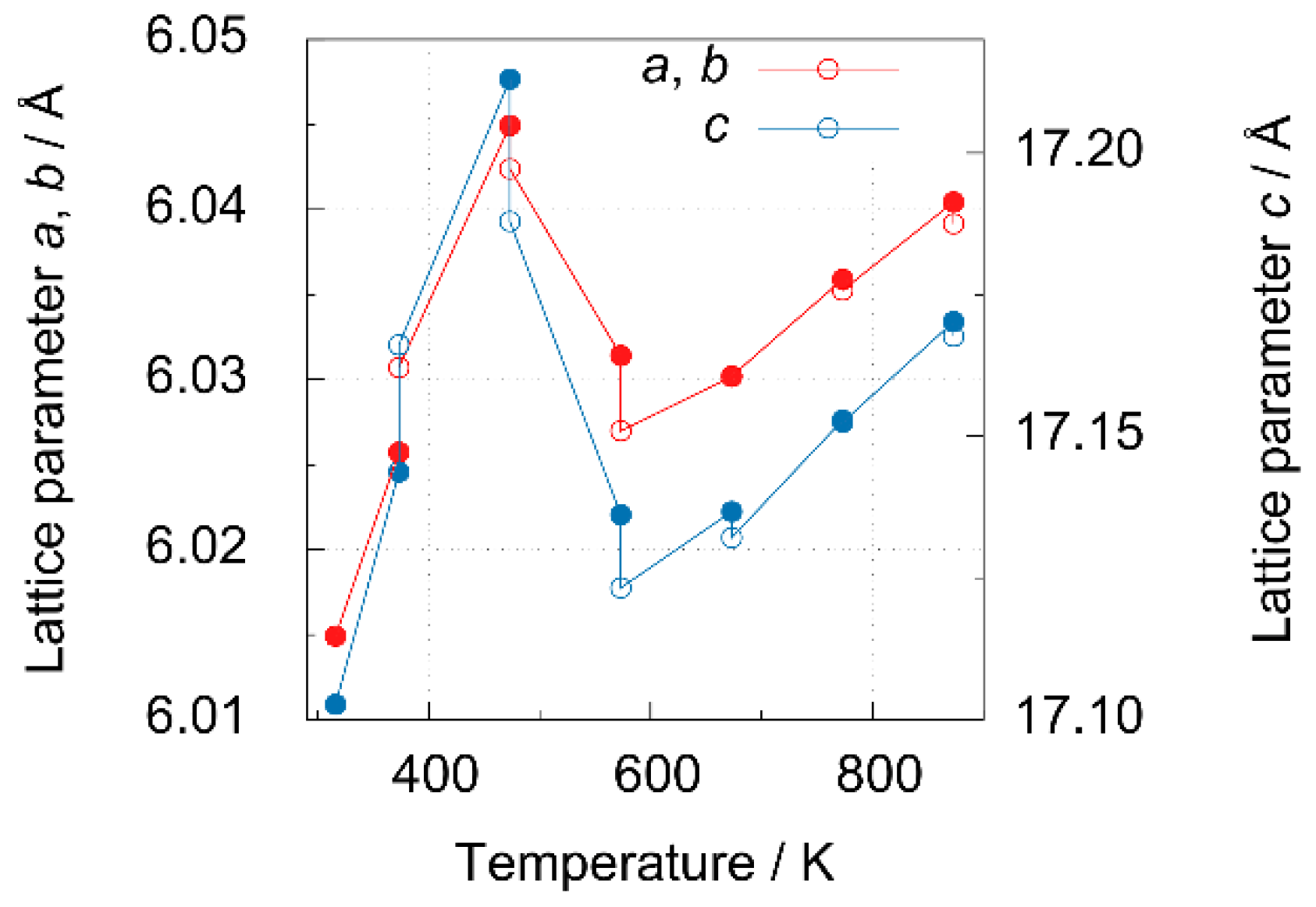
3.3.1. High-Temperature Neutron Diffraction Experiments at E6
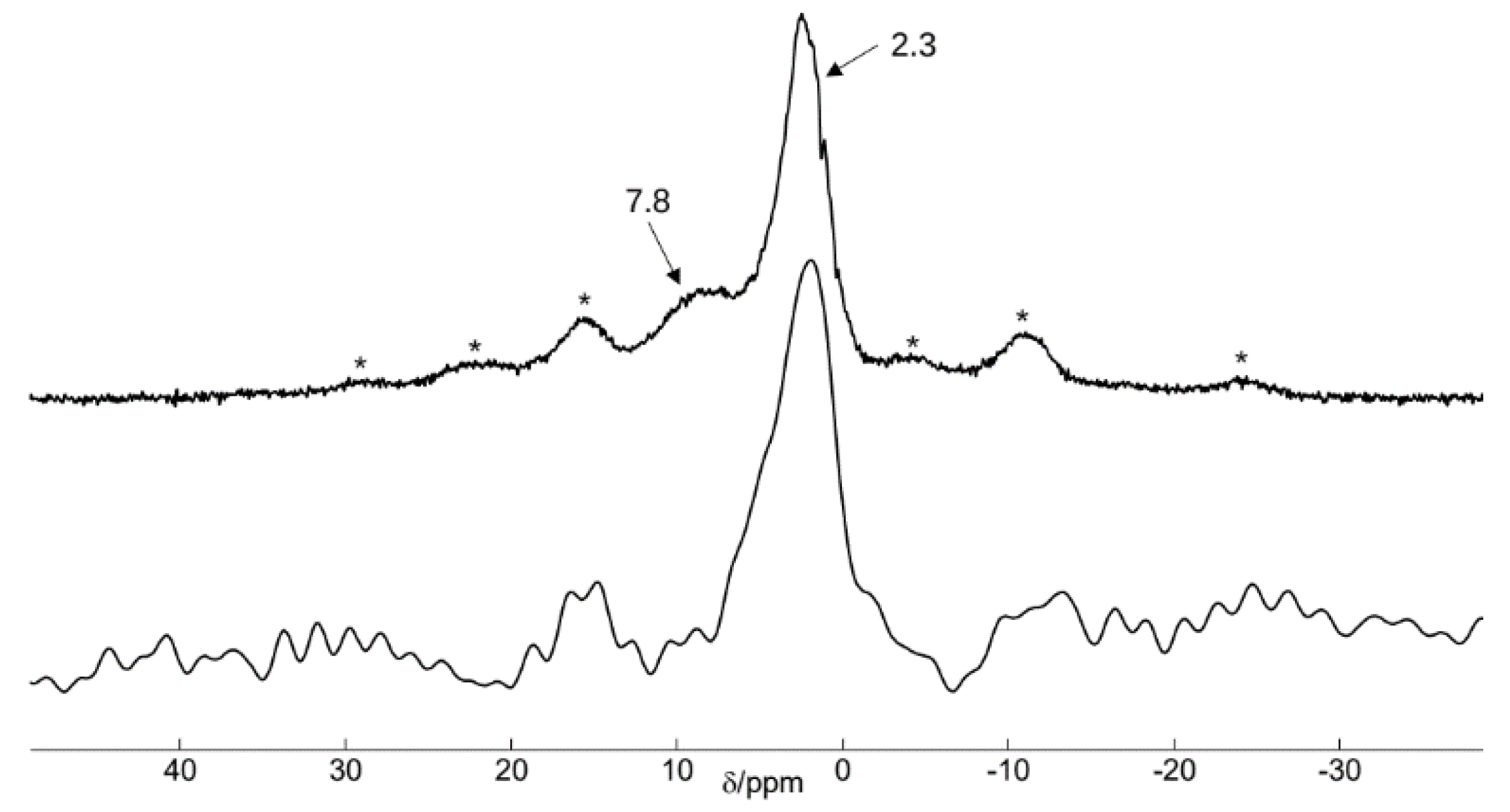
3.3.2. Nuclear Magnetic Resonance Spectroscopy
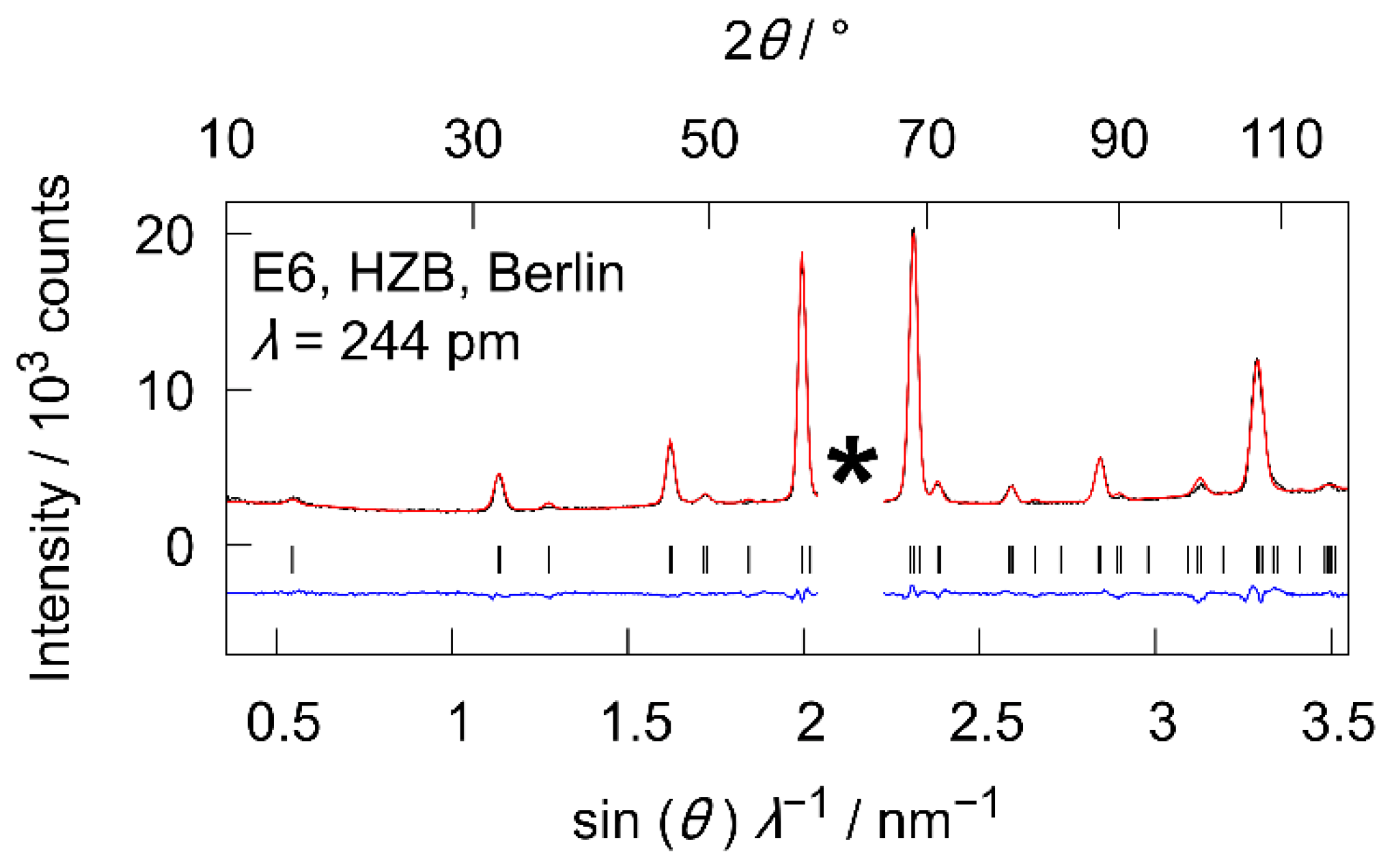
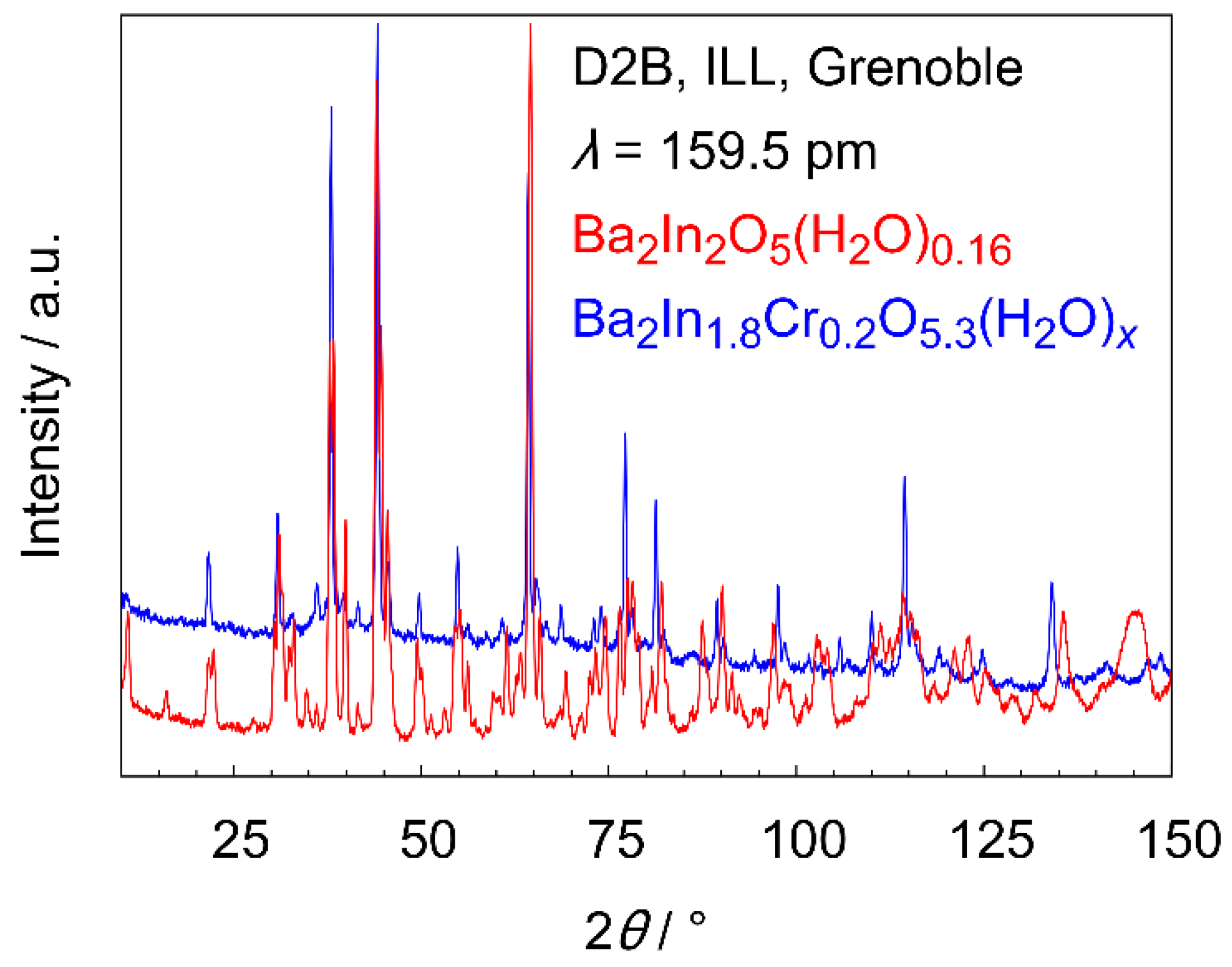
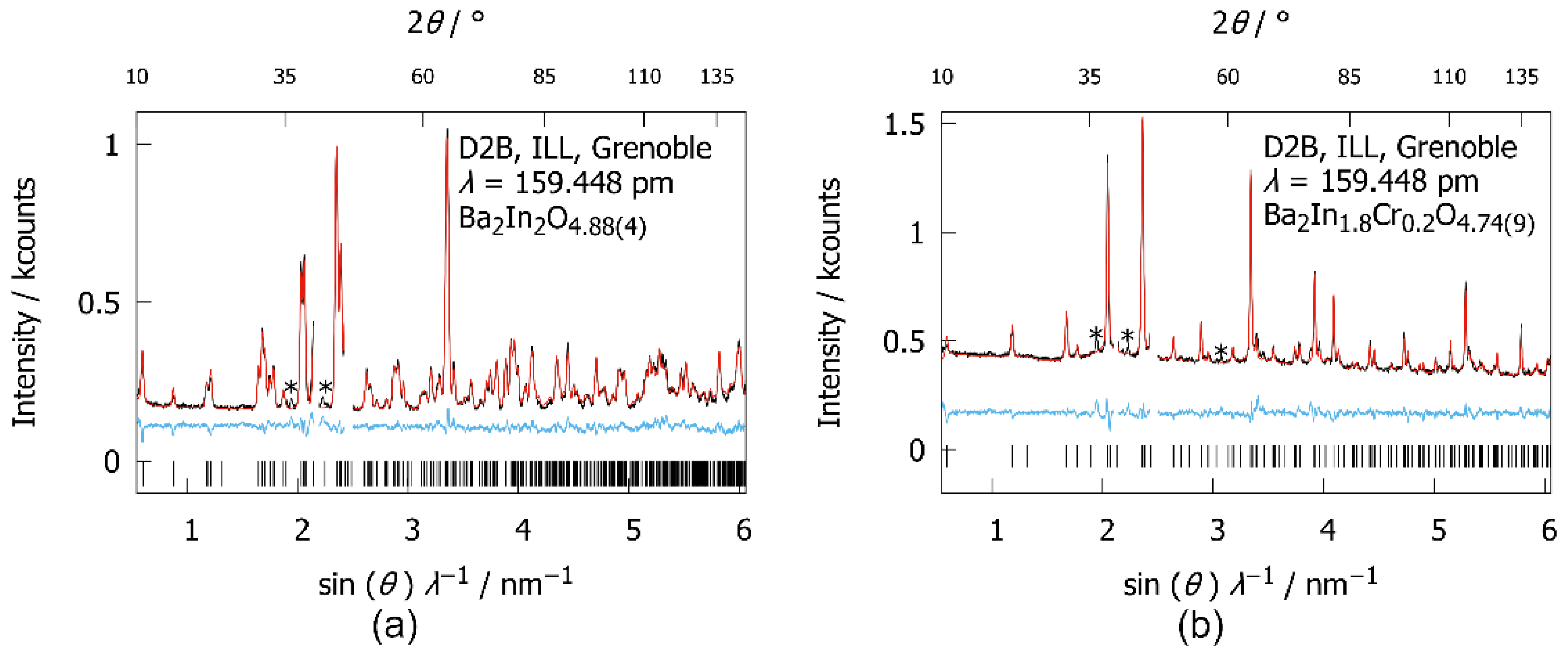
3.3.3. Low Temperature Powder Neutron Diffraction
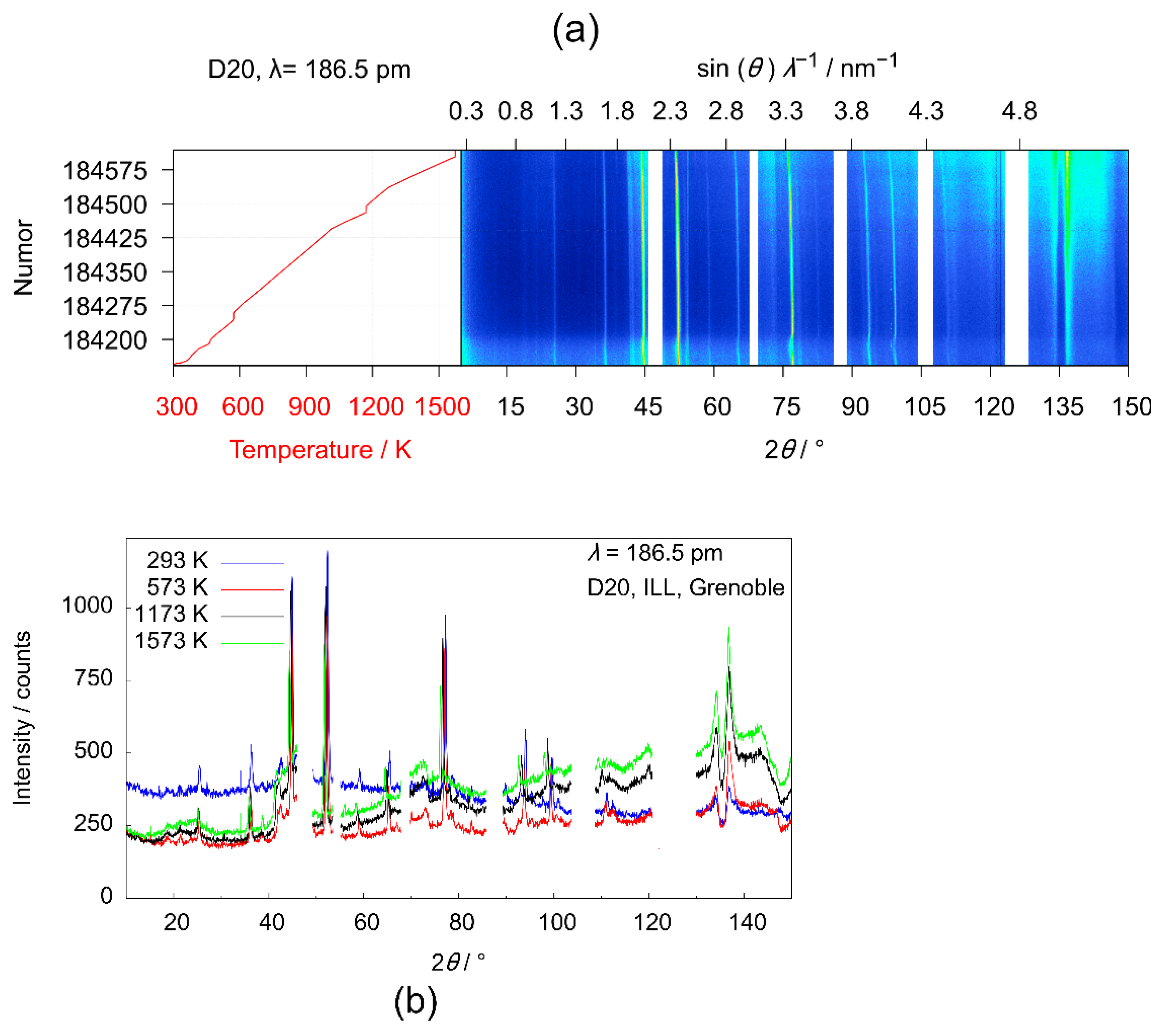
3.3.4. High-Temperature Powder Neutron Diffraction Experiments on D20
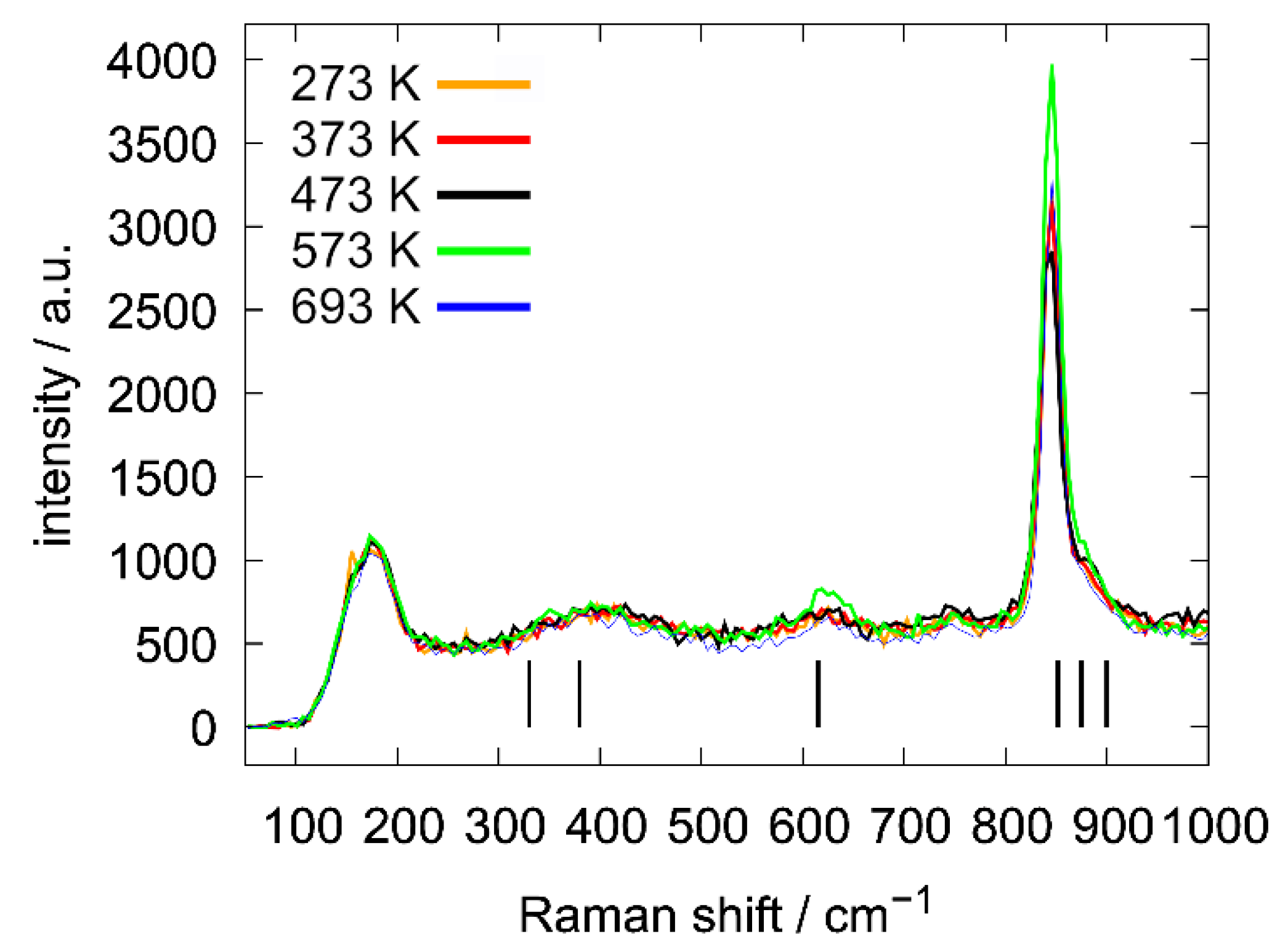
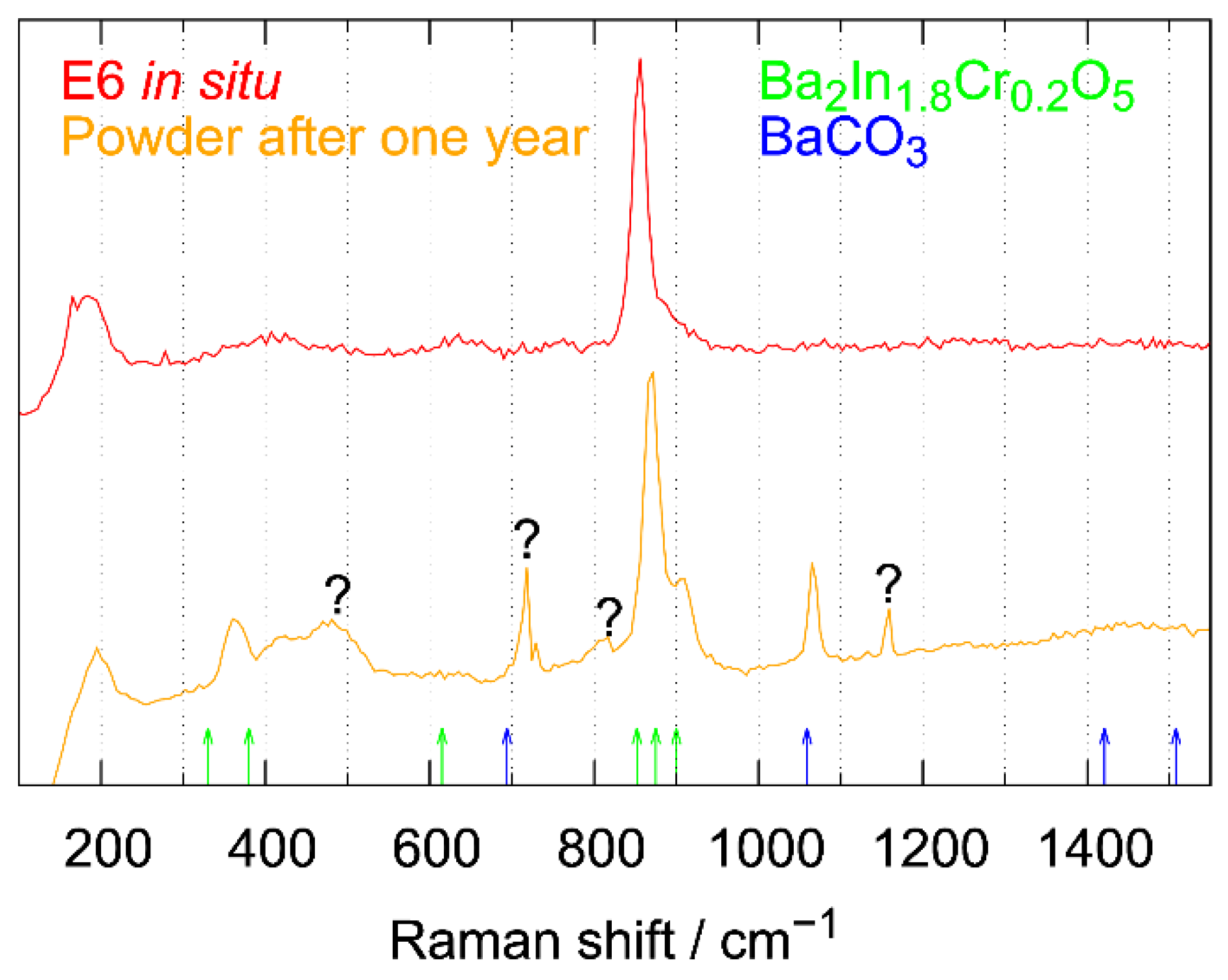
3.3.5. Powder Neutron Diffraction and Raman Spectroscopic Measurements
4. Discussion
4.1. X-ray Diffraction
4.2. Low-Temperature Neutron Diffraction
4.3. High-Temperature Neutron Diffraction
4.4. Nuclear Magnetic Resonance Spectroscopy
5. Conclusion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yoon, S.; Nikoee, S.; Ranjbar, M.; Ziegenbalg, D.; Widenmeyer, M.; Weidenkaff, A. Strongly affected photocatalytic CO2 reduction by CO2 adsorbed to the surface of Ba2(In1.8Cr0.2)O5·(H2O)δ powders. Solid State Sci. 2020, 105, 106212. [Google Scholar] [CrossRef]
- Yoon, S.; Gaul, M.; Sharma, S.; Son, K.; Hagemann, H.; Ziegenbalg, D.; Schwingenschlogl, U.; Widenmeyer, M.; Weidenkaff, A. Photocatalytic CO2 reduction by Cr-substituted Ba2(In2-xCrx)O5·(H2O)δ(0.04 ≤ x ≤ 0.60). Solid State Sci. 2018, 78, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.; Son, K.; Hagemann, H.; Widenmeyer, M.; Weidenkaff, A. Cr-substitution in Ba2In2O5·(H2O)x (x = 0.16, 0.74). Solid State Sci. 2017, 73, 1–6. [Google Scholar] [CrossRef]
- Widenmeyer, M.; Wiegers, K.-S.; Chen, G.; Yoon, S.; Feldhoff, A.; Weidenkaff, A. Engineering of oxygen pathways for better oxygen permeability in Cr-substituted Ba2In2O5 membranes. J. Membr. Sci. 2020, 595, 117558. [Google Scholar] [CrossRef]
- Zhang, G. Protonic conduction in Ba2In2O5. Solid State Ion. 1995, 82, 153–160. [Google Scholar] [CrossRef]
- Bielecki, J.; Parker, S.F.; Ekanayake, D.; Rahman, S.M.H.; Börjesson, L.; Karlsson, M. Short-range structure of the brownmillerite-type oxide Ba2In2O5 and its hydrated proton-conducting form BaInO3H. J. Mater. Chem. A 2014, 2, 16915–16924. [Google Scholar] [CrossRef]
- Perrichon, A.; Jiménez-Ruiz, M.; Mazzei, L.; Rahman, S.M.H.; Karlsson, M. Local structure and vibrational dynamics of proton conducting Ba2In2O5(H2O)x. J. Mater. Chem. A 2019, 7, 17626–17636. [Google Scholar] [CrossRef] [Green Version]
- Mazzei, L.; Piccinelli, F.; Bettinelli, M.; Parker, S.F.; Karlsson, M. The effect of cation substitution on the local coordination of protons in Ba2In1.85M0.15O6H2 (M = In, Ga, Sc and Y). Solid State Ion. 2021, 365, 115624. [Google Scholar] [CrossRef]
- Tyagi, D.; Shirsat, A.N.; Saha, B.; Varma, S. Phase stabilisation, chemical behaviour and protonic conductivity of vanadium doped Barium Indate for ITSOFC application. J. Alloys Compd. 2021, 877, 160298. [Google Scholar] [CrossRef]
- Bielecki, J.; Parker, S.F.; Mazzei, L.; Börjesson, L.; Karlsson, M. Structure and dehydration mechanism of the proton conducting oxide Ba2In2O5(H2O)x. J. Mater. Chem. A 2016, 4, 1224–1232. [Google Scholar] [CrossRef] [Green Version]
- Schober, T.; Friedrich, J.; Krug, F. Phase transition in the oxygen and proton conductor Ba2In2O5 in humid atmospheres below 300 °C. Solid State Ion. 1997, 99, 9–13. [Google Scholar] [CrossRef]
- Goodenough, J.; Manthiram, A.; Paranthaman, P.; Zhen, Y. Fast oxide-ion conduction in intergrowth structures. Solid State Ion. 1992, 52, 105–109. [Google Scholar] [CrossRef]
- Hashimoto, T.; Ueda, Y.; Yoshinaga, M.; Komazaki, K.; Asaoka, K.; Wang, S. Observation of Two Kinds of Structural Phase Transitions in the Ba2In2O5 System. J. Electrochem. Soc. 2002, 149, A1381. [Google Scholar] [CrossRef]
- Speakman, S. In-situ diffraction study of Ba2In2O5. Solid State Ion. 2002, 149, 247–259. [Google Scholar] [CrossRef]
- Lei, H.; Zhou, Y.; Jiang, P.; Wang, W.; Cao, W.; Xue, Y. Synthesis and optical properties of Cr-doped Ba2In2O5 orange-brown oxides. Ceram. Int. 2018, 44, 19751–19755. [Google Scholar] [CrossRef]
- Jayaraman, V.; Magrez, A.; Caldes, M.; Joubert, O.; Taulelle, F.; Rodriguez-Carvajal, J.; Piffard, Y.; Brohan, L. Characterization of perovskite systems derived from Ba2In2O5 Part II: The proton compounds Ba2In2(1−x)Ti2xO4+2x(OH)y [0 ≤ x ≤ 1; y ≤ 2(1 − x)]. Solid State Ion. 2004, 170, 25–32. [Google Scholar] [CrossRef]
- Martinez, J.-R.; Mohn, C.E.; Stølen, S.; Allan, N.L. Ba2In2O4(OH)2: Proton sites, disorder and vibrational properties. J. Solid State Chem. 2007, 180, 3388–3392. [Google Scholar] [CrossRef]
- Dervişoğlu, R.; Middlemiss, D.S.; Blanc, F.; Lee, Y.-L.; Morgan, D.; Grey, C.P. Joint Experimental and Computational 17O and 1H Solid State NMR Study of Ba2In2O4(OH)2 Structure and Dynamics. Chem. Mater. 2015, 27, 3861–3873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widenmeyer, M.; Hansen, T.C.; Meissner, E.; Niewa, R. Formation and Decomposition of Iron Nitrides Observed by in situ Powder Neutron Diffraction and Thermal Analysis. Z. Anorg. Allg. Chem. 2014, 640, 1265–1274. [Google Scholar] [CrossRef]
- Widenmeyer, M.; Hansen, T.C.; Leineweber, A.; Weidenkaff, A.; Niewa, R. Nitrogen Transfer between Solid Phases in the System Mn-N Detected via in situ Neutron Diffraction. Z. Anorg. Allg. Chem. 2017, 643, 1929–1938. [Google Scholar] [CrossRef] [Green Version]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Buchsteiner, A.; Stüßer, N. Optimizations in angular dispersive neutron powder diffraction using divergent beam geometries. Nucl. Instrum. Methods Phys. Res. Sect. A 2009, 598, 534–541. [Google Scholar] [CrossRef]
- Hansen, T.C.; Henry, P.F.; Fischer, H.E.; Torregrossa, J.; Convert, P. The D20 instrument at the ILL: A versatile high-intensity two-axis neutron diffractometer. Meas. Sci. Technol. 2008, 19, 34001. [Google Scholar] [CrossRef]
- Finger, R.; Hansen, T.C.; Kohlmann, H. Validation of a Sapphire Gas-Pressure Cell for Real-Time In Situ Neutron Diffraction Studies of Hydrogenation Reactions. Quantum Beam Sci. 2021, 5, 22. [Google Scholar] [CrossRef]
- Widenmeyer, M.; Niewa, R.; Hansen, T.C.; Kohlmann, H. In situ Neutron Diffraction as a Probe on Formation and Decomposition of Nitrides and Hydrides: A Case Study. Z. Anorg. Allg. Chem. 2013, 639, 285–295. [Google Scholar] [CrossRef]
- Robin Bendall, M.; Gordon, R.E. Depth and refocusing pulses designed for multipulse NMR with surface coils. J. Magn. Reson. (1969) 1983, 53, 365–385. [Google Scholar] [CrossRef]
- Feike, M.; Demco, D.E.; Graf, R.; Gottwald, J.; Hafner, S.; Spiess, H.W. Broadband Multiple-Quantum NMR Spectroscopy. J. Magn. Reson. Ser. A 1996, 122, 214–221. [Google Scholar] [CrossRef]
- Rodríguez-Carvajal, J. Recent advances in magnetic structure determination by neutron powder diffraction. Phys. B Condens. Matter 1993, 192, 55–69. [Google Scholar] [CrossRef]
- Massiot, D.; Fayon, F.; Capron, M.; King, I.; Le Calvé, S.; Alonso, B.; Durand, J.-O.; Bujoli, B.; Gan, Z.; Hoatson, G. Modelling one- and two-dimensional solid-state NMR spectra. Magn. Reson. Chem. 2002, 40, 70–76. [Google Scholar] [CrossRef]
- Boyd, S.; Wright, I.P.; Pillinger, C.T. Stepped-heating of carbonates and carbon-bearing quartz grains. Chem. Geol. 1997, 134, 303–310. [Google Scholar] [CrossRef]
- Sears, V.F. Neutron scattering lengths and cross sections. Neutron News 1992, 3, 26–37. [Google Scholar] [CrossRef]
- Pasierb, P.; Komornicki, S.; Rokita, M.; Rekas, M. Structural properties of Li2CO3–BaCO3 system derived from IR and Raman spectroscopy. J. Mol. Struct. 2001, 596, 151–156. [Google Scholar] [CrossRef]
- Zaki, M.I.; Fahim, R.B. Thermal decomposition and creation of reactive solid surfaces. J. Therm. Anal. Calorim. 1986, 31, 825–834. [Google Scholar] [CrossRef]
- Jóźwiak, W.K.; Ignaczak, W.; Dominiak, D.; Maniecki, T.P. Thermal stability of bulk and silica supported chromium trioxide. Appl. Catal. A Gen. 2004, 258, 33–45. [Google Scholar] [CrossRef]
- Liu, B.; Fang, Y.; Terano, M. High resolution X-ray photoelectron spectroscopic analysis of transformation of surface chromium species on Phillips CrOx/SiO2 catalysts isothermally calcined at various temperatures. J. Mol. Catal. A Chem. 2004, 219, 165–173. [Google Scholar] [CrossRef]
- Holleman, A.F.; Wiberg, E. Lehrbuch der Anorganischen Chemie, 101st ed.; Walter de Gruyter: Berlin, Germany, 1995. [Google Scholar]
- Hewston, T.A.; Chamberland, B.L. Magnetic and structural studies of Cr2O5 and Cr3O8. J. Magn. Magn. Mater. 1984, 43, 89–95. [Google Scholar] [CrossRef]
- Chamberland, B.L. The chemical and physical properties of CrO2 and tetravalent chromium oxide derivatives. Crit. Rev. Solid State Mater. Sci. 1977, 7, 1–31. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Measurement Program | Heating Rate (K·min−1) | Tmax (K) | Cooling Rate (K·min−1) | Gas Type and Flow Rate |
|---|---|---|---|---|
| 1 | 10 | 1473 | 10 | 60 sccm Ar |
| 2 | 10 | 1473 | – | 60 sccm Ar |
| 3 | 10 | 1473 | 10 | 60 sccm Air |
| 4 | 10 | 1473 | 1 | 60 sccm Air |
| 5 | 2 | 1473 | 2 | 60 sccm N2 |
| T(K) | Measurement Program | Δm(Ba2In2O5(H2O)0.16) | Δm(Ba2In1.8Cr0.2O5.3(H2O)x) | ||
|---|---|---|---|---|---|
| 800 | 1; Ar, 10 K·min−1 | −0.60% * | −0.61% [3] | −2.18% * | −2.19% [3] |
| 1473 | 1; Ar, 10 K·min−1 | −0.82% * | −1.02% [3] | −2.82% * | −2.81% [3] |
| 800 | 2; Ar, 10 K·min−1 | N/A | N/A | −2.28% * | −2.19% [3] |
| 1473 | 2; Ar, 10 K·min−1 | N/A | N/A | −2.95% * | −2.81% [3] |
| 800 | 3; Air, 10 K·min−1 | N/A | N/A | −2.23% * | N/A |
| 1473 | 3; Air, 10 K·min−1 | N/A | N/A | −2.94% * | N/A |
| 800 # | 3; Air, 10 K·min−1 | N/A | N/A | +0.18% * | N/A |
| 800 | 4; Air, 10 K·min−1 | N/A | N/A | −2.20% * | N/A |
| 1473 | 4; Air, 10 K·min−1 | N/A | N/A | −2.90% * | N/A |
| 800 # | 4; Air, 1 K·min−1 | N/A | N/A | +0.16% * | N/A |
| 800 | 5; N2, 2 K·min−1 | N/A | N/A | −2.24% * | N/A |
| 1473 | 5; N2, 2 K·min−1 | N/A | N/A | −2.90% * | N/A |
| Temperature/K | Oxygen Content |
|---|---|
| 293 | 5.32(4) |
| 573 | 5.36(4) |
| 1173 | 5.26(4) |
| 1573 | 5.08(6) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Finger, R.; Widenmeyer, M.; Hansen, T.C.; Wallacher, D.; Savvin, S.; Bertmer, M.; Weidenkaff, A.; Kohlmann, H. Effects of Cr Doping and Water Content on the Crystal Structure Transitions of Ba2In2O5. Crystals 2021, 11, 1548. https://doi.org/10.3390/cryst11121548
Finger R, Widenmeyer M, Hansen TC, Wallacher D, Savvin S, Bertmer M, Weidenkaff A, Kohlmann H. Effects of Cr Doping and Water Content on the Crystal Structure Transitions of Ba2In2O5. Crystals. 2021; 11(12):1548. https://doi.org/10.3390/cryst11121548
Chicago/Turabian StyleFinger, Raphael, Marc Widenmeyer, Thomas C. Hansen, Dirk Wallacher, Stanislav Savvin, Marko Bertmer, Anke Weidenkaff, and Holger Kohlmann. 2021. "Effects of Cr Doping and Water Content on the Crystal Structure Transitions of Ba2In2O5" Crystals 11, no. 12: 1548. https://doi.org/10.3390/cryst11121548
APA StyleFinger, R., Widenmeyer, M., Hansen, T. C., Wallacher, D., Savvin, S., Bertmer, M., Weidenkaff, A., & Kohlmann, H. (2021). Effects of Cr Doping and Water Content on the Crystal Structure Transitions of Ba2In2O5. Crystals, 11(12), 1548. https://doi.org/10.3390/cryst11121548