Abstract
Thiophene and sulfonamide derivatives serve as biologically active compounds, used for the manufacture of large numbers of new drugs. In this study, 11 selected derivatives of thiophene sulfonamide were computed for their geometric parameters, such as hyperpolarizability, chemical hardness (ƞ), electronic chemical potential (μ), electrophilicity index (ω), ionization potential (I), and electron affinity (A). In addition, FT-IR and UV-Vis spectra were also simulated through theoretical calculations. The geometrical parameters and vibrational frequencies with assignments of the vibrational spectra strongly resemble the experimentally calculated values. Besides, the frontier molecular orbitals were also determined for various intramolecular interactions that are responsible for the stability of the compounds. The isodensity surfaces of the frontier molecular orbitals (FMOs) are the same pattern in most of the compounds, but in some compounds are disturbed due to the presence of highly electronegative hetero-atoms. In this series of compounds, 3 shows the highest HOMO–LUMO energy gap and lowest hyperpolarizability, which leads to the most stable compound and less response to nonlinear optical (NLO), while 7 shows the lowest HOMO–LUMO energy gap and highest hyperpolarizability, which leads to a less stable compound and a high NLO response. All compounds have their extended three-dimensional p-electronic delocalization which plays an important role in studying NLO responses.
1. Introduction
Thiophene is a stable compound that is readily available in the market, and the investigation of thiophene and its compounds has been a continual matter [1]. They are used in organic cells and also act as organic light-emitting diodes (OLEDs), organic field-effect transistors (OFETs) [2], etc. Fused thiophenes are electron-rich structures that enable them to act as good electron donors, thus finding applications in organic semiconductors such as dye-sensitized solar cells [3,4]. Thiophenes have also found applications in sensing bisulfite, which is an environmental pollutant [5]. Heterocyclic compounds such as thiophene show biological activities in pharmaceutical and medicinal fields [6]. Derivatives of thiophene are used as biologically active molecules for different diseases [7]. Thiophene derivatives are important compounds for the discovery of new medicines. Substituted thiophenes show good antiproliferative [8], antioxidant [9], antimicrobial [10], and anticancer activity [11]. Imidazole coupled with thiophene acts as an antimycobacterial agent [12]. Thiophene-2-carboxylic acid has good agonistic activity against the GPR35 [13]. Thiazole coupled with thiophene acts as an adenosine receptor antagonist [14]. Among the large number of pharmaceutical compounds, the sulfonamide functional group plays a basic role in the manufacture of many drugs [15]. Sulfonamides are an important class of compound which produce various biologically active molecules [16]. Some important applications of sulfonamides are their therapeutic action against several bacterial species, most commonly positive and negative Gram-staining species. Sulfonamides and related moieties have been known to possess moderate to good activities against these microbes, and this has resulted in the synthesis of various important medicines of these compounds against various diseases caused by bacteria and other microbial species [17]. Sulfonamide derivatives are usually attached to the dihydropteroate synthase, which is used to catalyze folic acid metabolic pathways in bacteria and some eukaryotic cells [18].
Over the past two decades, density functional theory (DFT) has become an abundantly useful technique in many fields of chemistry [19]. Many experimental studies in inorganic and organic chemistry normally include such calculations, using a standard functional approximation, a standard basis, and a popular code [20]. The history of DFT starts from Dirac’s local exchange approximation and Thomas Fermi’s theory [21]. DFT seems to be an extremely successful approach for the description of the ground-state properties of metals, semiconductors, and insulators [22]. DFT investigations were accompanied not only to explain the structural properties but also to compare the theoretical structural parameters with those from X-ray diffraction outcomes [23]. Our research group already reported the synthesis and biological activities of various thiophene based molecules [24,25,26]. On behalf of the reported biological activities of thiophene derivatives, we decided on the computational study of selected thiophene sulfonamide derivatives (1–11).
2. Computational Details
This research was done using Gaussian 09W [27] to find the chemical and thermodynamic properties of molecules using DFT calculations. These calculations were performed using Becke–Lee–Yang–Parr’s three-parameter hybrid functional (B3LYP) [28,29] with Pople’s triple zeta basis set 6-311G(d,p). The solvent effects were added through the SMD parameter set [30] as implemented in Gaussian. The solvent used in all the calculations was 1,4-dioxane [31]. GaussView has been used to visualize the results of quantum mechanical calculations. It has been previously verified that the B3LYP functional can provide an excellent compromise between the accuracy of the vibrational spectra of large and medium-sized molecules and the computational efficiency [1]. A charge distribution diagram of frontier molecular orbitals was obtained which shows various properties of molecules, such as the energy of the highest occupied molecular orbital (EHOMO) and the energy of the lowest unoccupied molecular orbital (ELUMO), as well as global reactivity parameters such as chemical potential (µ), electrophilicity index (ω), and chemical hardness (η) to analyze the reactivity of the inhibitor molecules [9]. Reactivity descriptors such as ionization potential and electron affinity are calculated from HOMO and LUMO and chemical hardness (ƞ), and electronic chemical potential (μ) is calculated from ionization potential (I) and electron affinity (A). UV-Vis spectra were simulated using the time-dependent density functional theory (TD-DFT) [32] at the same level of theory as optimizations. The hyperpolarizabilities were calculated using the “polar” keyword as implemented in Gaussian 09. GaussSum [33] was used to draw the IR and UV-Vis spectra. The structures with the IUPAC names of the thiophene sulfonamide derivatives [26] under study are given in Figure S1 (see supplementary materials).
3. Results and Discussion
3.1. Geometrical Parameters
The geometrical parameters such as bond length and bond angle were calculated by applying the B3LYP/6-311G (d,p)/SMD1,4-dioxane level of theory. The bond lengths and bond angles for selected bonds of 1 to 11 are given in Table 1 and Table 2. The calculated intramolecular distances of S1–C2 and C5–S1 are in the range of 1.73 Å to 1.75 Å which deviate significantly from the experimental value of 2.74 Å [34]. The intramolecular distances of S=O and S–NH2 in the sulfonamide group lie from 1.45 Å to 1.46 Å and 1.67 Å to 1.68 Å, respectively, but the bond length of S=O in most of the compounds is 1.46 Å. The intramolecular distances S1–C2 and C5–S1 were calculated to be in the range of 1.73 Å to 1.75 Å. The experimentally calculated bond lengths of S=O and S–NH2 in the sulfonamide group in literature are 1.42 Å and 1.64 Å, respectively. The bond angles of S1–C2–C3 and C4–C5–S1 were calculated to be in the range of 110.84–112.44° and 110.63–112.45° respectively. The bond angles of O=S–NH2 and O=S=O were calculated to be in the range of 105.04–111.26° and 120.46–121.18°, which are very close to the experimental values 105.4° and 123.1° [35]. The dihedral angles between the rings in the molecules under study have been given in Table 3. All the optimized structures of selected thiophene sulfonamide derivatives under study are given in Figure 1.

Table 1.
Bond length Å of selected bonds of 1 to 11.
Table 2.
Bond angles (°) of selected bonds of 1 to 11.
Table 3.
Dihedral angles (°) of selected dihedrals of 1 to 11.
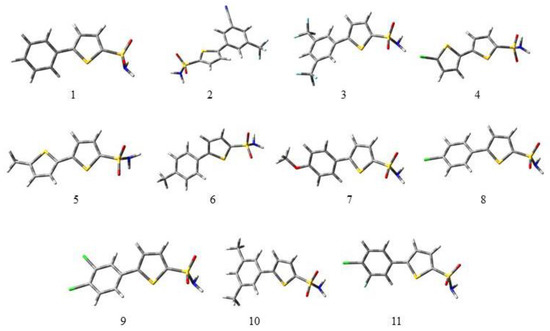
Figure 1.
Optimized geometries of all the derivatives (1 to 11) under study at the B3LYP/6-311G (d,p)/SMD1,4-dioxane level of theory. In 3D models, the grey color symbolizes carbon, white symbolizes hydrogen, the red color is for oxygen, green is for chlorine, the yellow color symbolizes sulfur, and the light blue color indicates fluorine.
3.2. Frontier Molecular Orbital Analysis
The frontier molecular orbitals (FMOs) are also used to describe the interactions of molecules with the other molecules [36]. These orbitals are the major aspect that tells us about the stability of the compounds. From the value of EHOMO we can determine the electron-donating ability, and from the value of ELUMO we find the electron-accepting ability. In conjugated systems there is a slight difference between HOMO–LUMO, due to which electron (charge) can transfer from one atom (electron donor groups) to the other (e-accepting groups) via a pi-conjugated system [37].
The iso-density surface of the FMOs showed almost the same style for all derivatives except 2, 3, 4, 7, and 8 due to the presence of electron-withdrawing groups (CF3, CN, Cl, F, CH3O) bonded to the substituted phenyl ring. The electronic cloud was spread over the whole compound, mostly on the rings (phenyl thiophene). The FMOs of 1 to 11 were obtained from optimized geometries at the B3LYP/6-311G (d,p)/SMD1,4-dioxane level of theory and are given in Figure 2.

Figure 2.
Frontier molecular orbitals of all the compounds under study calculated at the B3LYP/6-311G (d,p)/SMD1,4-dioxane level of theory.
The energy of HOMO–LUMO, the energy difference (∆E) in HOMO–LUMO, and the hyperpolarizability of thiophene sulfonamide derivatives are given in Table 4. The values of energy difference (∆E) of all of the thiophene sulfonamide compounds under study were analyzed in the range 4.65–3.44 eV. These values reflect that all the compounds are stable in the 1,4-dioxane solvent. As expected, the hyperpolarizability varies roughly as the inverse of the squared HOMO–LUMO gap (from the 2-state model).
Table 4.
Energies of HOMO–LUMO, energy gap, and hyperpolarizability values of 1 to 11. The units of βo are Hartree while all the other energies are shown in eV.
Compounds 3 and 1 have the highest energy gap (∆E) values among the thiophene sulfonamide series. This energy gap makes them the most stable compounds compared to the others. On the other hand, compound 7 has the lowest energy gap (∆E) value among the thiophene sulfonamide series. This lowest energy gap makes it the least stable compound in the series.
The energy gap decreased in the following order.
3 > 1 > 2 > 10 > 8 > 11 = 6 > 9 > 4 > 5 > 7
3.3. Nonlinear Optical (NLO) Properties
Over the past several decades, the design of new compounds with large nonlinear optical (NLO) responses has attracted extensive theoretical and experimental attention due to their large number of applications in electro-optical and optical devices [38]. The significant requirements for becoming a high-quality NLO material include a high optical coefficient, high thermal and chemical stability, and good transparency [39]. In the thiophene sulfonamide series, molecule 7 with substituted methoxy groups (OCH3) at the para position has the highest hyperpolarizability value, and molecule 3 with a CF3– group at the meta position has the lowest hyperpolarizability value.
The hyperpolarizability decreases in the following order.
7 > 5 > 4 > 6 > 8 > 11 > 10 > 9 > 2 > 1 > 3
3.4. Molecular Electrostatic Potential (MESP)
The molecular electrostatic potential (MESP) provides information about the size, charge, and overall shape of the molecule [40]. It is a very useful tool to analyze the relationship between the molecular structure and the physicochemical properties of compounds including drugs and biomolecules [41]. In the MESP map, the different values of the electrostatic potential at the surface are represented by different colors. The most negative electrostatic potential region is illustrated by red color, the most positive electrostatic potential region is represented by blue color, and the green color indicates the zero potential regions [42]. The MESP surfaces of all the compounds under study are shown in Figure 3. The most negative electrostatic potential region is mainly located on the oxygen of the carboxyl group as a probable position for the attack of electron-deficient species, while the largest electrostatic potential region is located on the hydrogen of the NH2 group as a potential site for the attack of electron-rich species.
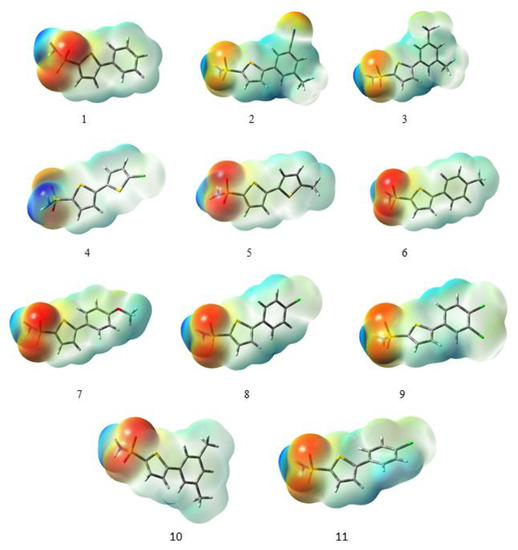
Figure 3.
Molecular electrostatic potentials maps of all derivatives under study calculated at the B3LYP/6-311G (d,p)/SMD1,4-dioxane level of theory.
3.5. IR Spectra
Infrared or vibrational spectroscopy is an effective technique for identifying functional groups in organic compounds [43]. The IR spectra of the thiophene sulfonamide derivatives under study have not been studied in detail experimentally or theoretically in the literature. We calculated the theoretical IR spectra of thiophene sulfonamide derivatives at the B3LYP/6-311G (d,p)/SMD1,4-dioxane level of theory.
The vibrational investigation aims to find the vibrational modes associated with specific molecular structures of calculated compounds (Table 5). The infrared spectra of 1 to 11 are given in Figure 4. None of the calculated vibrational spectra have an imaginary frequency that confirms the structures as true minima.
Table 5.
Calculated frequency ranges and vibrational modes based on them for compounds 1–11.
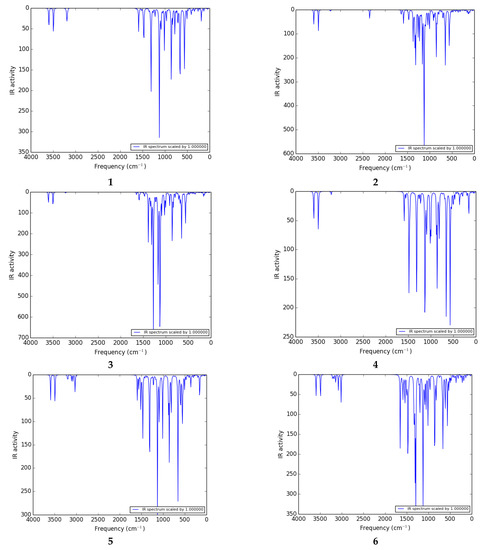
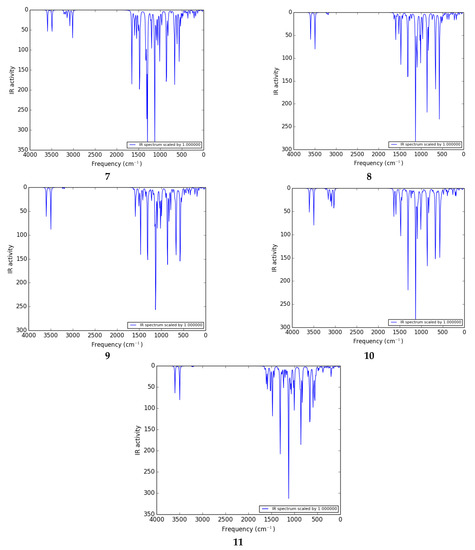
Figure 4.
IR spectra of 1 to 11 calculated at the B3LYP/6-311G (d,p)/SMD1,4-dioxane level of theory.
H–N–H vibrations: The thiophene sulfonamide derivatives have a large number of vibrations. The NH2 group gives rise to one of the most readily recognizable bands in the IR spectrum. The highest frequency range 3590–3610 cm−1 belongs to the deformation (out of plane) stretch of the NH2 group in most of the compounds. The H–N–H scissoring and wagging vibration appears as bands at 3602 and 3597 cm−1 as shown in the spectra of 3 and 2 respectively. The H–N–H antisymmetric stretching vibration appears in the range of 3590–3600 cm−1 as shown in the spectra of 1, 4, and 5. In 2, the amino group shows one absorption band at 3494 cm−1 arising from twisting vibrations. The H–N–H wagging, scissoring, and deformation vibrations are observed in the range of 1580–1600 cm−1. The H–N–H antisymmetric stretching vibrations are observed in the ranges of 1580–1600, 850–865, and 655–690 cm−1.
C–H vibrations: The C–H deformation and stretching vibrations in the thiophene ring are observed in the ranges of 3220–3235, 1220–1265, 920–940 cm−1, and 3210–3235, 1215–1265 cm−1, respectively. In the literature, the stretching vibrations of C–H of the aromatic molecules arise in the range of 2900–3150 cm−1 [44] which are in good agreement with the C–H deformation, and stretching vibrations in the phenyl ring are observed in the ranges of 3150–3210, 1180–1205, 820–975 cm−1 and 3165–3200, 1120–1210 cm−1, respectively.
Ph–CN vibration: The Ph–CN deformation vibrations are observed at 3246 cm−1 in 2.
C–C vibration: The C–C stretching vibrations of thiophene and phenyl rings are observed in the ranges of 1590–1655 and 1550–1650 cm−1, respectively. In the literature, the experimentally calculated C–C stretching vibrations of thiophene and benzene rings appeared in the range of 1465.2–1597.3 cm−1 [43], which is in good agreement with the theoretically calculated frequency.
O=S=O vibration: The antisymmetric stretching vibrations of O=S=O are observed at 1308 cm−1 in 1, and deformation vibrations are observed in the range of 1300–1315 cm−1 in most of the compounds.
3.6. UV-Vis Spectrum
The UV-Vis spectrum is a photophysical property of molecules, caused by the absorption of light by electrons that are excited to higher energy levels [45]. When the absorption wavelength (λmax) of ultraviolet light is high, the tendency of thiophene derivatives to be easily oxidized can be considered as a basic parameter for predicting their reactivity, and this is a new method of calculating their kinetics, which can replace time-consuming and expensive experiments [46]. The calculated excitation energies (E), absorption wavelengths (λ), and oscillator strengths (f) are given in Table 6. The theoretical UV-Vis spectrum of 1 is given in Figure 5. The theoretical UV-Vis spectra of the remaining compounds are given in the supporting information (Figure S2).
Table 6.
Theoretical calculated excitation energies (E), absorption wavelengths (λ max), and oscillator strengths (f) of all compounds under study.
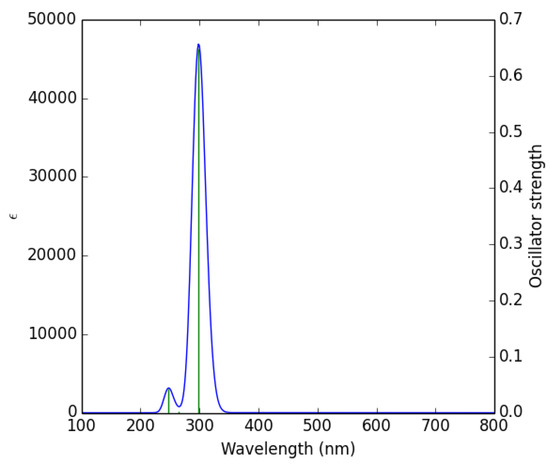
Figure 5.
Theoretically calculated UV-Vis spectrum of 1.
3.7. Conceptual DFT Reactivity Descriptors
The tendency of a chemical substance to react with another chemical substance can be defined as chemical reactivity. Reactivity descriptors such as chemical hardness (ƞ), electronic chemical potential (μ), electrophilicity index (ω), ionization potential, and electron affinity are calculated from the energies of HOMO and LUMO. Ionization potential and electron affinity are the negative of EHOMO and ELUMO according to Koopman’s theorem [47]; other thermodynamic properties, chemical hardness (ƞ), chemical softness (σ), electronic chemical potential (μ), and electrophilicity index (ω), can be calculated from these by using Equations (1)–(4) [48]. The ionization potential (I), electron affinity (A), chemical hardness (ƞ), chemical softness (σ), electronic chemical potential (μ), and electrophilicity index (ω) are given in Table 7.
η = (I − A)/2
μ = − (I + A)/2
ω = μ2/2η
σ = 1/η
Table 7.
The values of the important reactivity descriptors of compounds 1–11.
A higher energy of HOMO shows that a molecule will be more reactive in chemical reactions with electrophiles, with the corresponding lower LUMO energy for chemical reactions with nucleophiles [49]. The gap between these two (HOMO and LUMO) provides information about the stability of the compound in the given medium. The relatively larger gap between HOMO and LUMO is evidence of general stability of these compounds, which is in support of the reactivity descriptors shown in Table 7. The low chemical softness and higher hardness support this fact. The chemical potential (μ) is the tendency of an electron to escape from a stable molecule. The negative chemical potential means that the complex is stable and will not spontaneously decompose into elements. Hardness indicates the resistance to changes in the distribution of electrons in molecules [50].
4. Conclusions
The structural properties, thermodynamic properties, fundamental vibrational frequencies, and the optimized ground state geometries of the thiophene sulfonamide derivatives under study are reported for the first time by applying DFT calculations. The energy gap of HOMO and LUMO reveals the stability of the compounds in the given solution. Moreover, the nonlinear optical, first-order hyperpolarizability characteristics of the molecule explain that the molecule is an interesting agent for further studies of nonlinear optical characteristics. The chemical reactivity relations of these derivatives were determined through local reactivity descriptors such as electrophilicity, electronegativity, softness, hardness, and chemical potential, by using DFT methods.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4352/11/2/211/s1. Figure S1: Structures and names of all compounds under study; Figure S2: Theoretical calculated UV-Vis spectra of 2–11.
Author Contributions
Conceptualization, A.M. (Adeel Mubarik) and N.R.; methodology, N.R. and M.A.H., software, A.M. (Adeel Mubarik), N.R., M.A.H., A.M. (Asim Mansha), and M.Z.; formal analysis, M.R.S., M.A.F.S., and E.M.A.; investigation, A.M., N.R., M.A.H., A.M. (Asim Mansha), and M.Z.; resources, N.R.; data curation, A.M. (Adeel Mubarik) and N.R.; writing—original draft preparation, A.M. (Adeel Mubarik), N.R., M.A.H., A.M. (Asim Mansha), and M.Z.; writing—review and editing, A.M. (Adeel Mubarik), N.R., M.A.H., A.M. (Asim Mansha), M.Z., M.R.S., and A.A.; visualization, N.R.; supervision, N.R.; project administration, N.R.; funding acquisition, M.R.S.; All authors have read and agreed to the published version of the manuscript.
Funding
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding this work through Research Group no. RG-1441-453.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article or supplementary material.
Acknowledgments
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding this work through Research Group no. RG-1441-453.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Xiao-Hong, L.; Xiang-Ru, L.; Xian-Zhou, Z. Calculation of vibrational spectroscopic and NMR parameters of 2-dicyanovinyl-5-(4-N, N-dimethylaminophenyl) thiophene by ab initio HF and density functional methods. Comput. Chem. 2011, 969, 27–34. [Google Scholar] [CrossRef]
- Chen, Y.; Wan, X.; Long, G. High performance photovoltaic applications using solution-processed small molecules. Acc. Chem. Res. 2013, 46, 2645–2655. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhao, C.; Wang, H. Recent Progress in Synthesis and Application of Thiophene Oligomers Based on Bithiophene Dicarbanions. Chem. Rec. 2016, 16, 797–809. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Wenger, S.; Xu, M.; Gao, F.; Jing, X.; Wang, P.; Zakeeruddin, S.M.; Grätzel, M. An Organic Sensitizer with a Fused Dithienothiophene Unit for Efficient and Stable Dye-Sensitized Solar Cells. J. Am. Chem. Soc. 2008, 130, 9202–9203. [Google Scholar] [CrossRef]
- Chao, J.; Wang, X.; Liu, Y.; Zhang, Y.; Huo, F.; Yin, C.; Zhao, M.; Sun, J.; Xu, M. A pyrene-thiophene based fluorescent probe for ratiometric sensing of bisulfite and its application in vivo imaging. Sens. Actuator B Chem. 2018, 272, 195–202. [Google Scholar] [CrossRef]
- Sial, N.; Rasool, N.; Rizwan, K.; Altaf, A.A.; Ali, S.; Malik, A.; Zubair, M.; Akhtar, A.; Kausar, S.; Shah, S.A.A. Efficient synthesis of 2, 3-diarylbenzo [b] thiophene molecules through palladium (0) Suzuki–Miyaura cross-coupling reaction and their antithrombolyitc, biofilm inhibition, hemolytic potential and molecular docking studies. Med. Chem. Res. 2020, 29, 1486–1496. [Google Scholar] [CrossRef]
- Wan, Y.; Wallinder, C.; Plouffe, B.; Beaudry, H.; Mahalingam, A.; Wu, X.; Johansson, B.; Holm, M.; Botoros, M.; Karlén, A. Design, synthesis, and biological evaluation of the first selective nonpeptide AT2 receptor agonist. J. Med. Chem. 2004, 47, 5995–6008. [Google Scholar] [CrossRef]
- Do, C.V.; Faouzi, A.; Barette, C.; Farce, A.; Fauvarque, M.-O.; Colomb, E.; Catry, L.; Berthier-Vergnes, O.; Haftek, M.; Barret, R. Synthesis and biological evaluation of thiophene and benzo [b] thiophene analogs of combretastatin A-4 and isocombretastatin A-4: A comparison between the linkage positions of the 3, 4, 5-trimethoxystyrene unit. Bioorg. Med. Chem. Lett. 2016, 26, 174–180. [Google Scholar] [CrossRef]
- Rizwan, K.; Zubair, M.; Rasool, N.; Mahmood, T.; Ayub, K.; Alitheen, N.B.; Aziz, M.N.M.; Akhtar, M.N.; Bukhary, S.M.; Ahmad, V.U. Palladium (0) catalyzed Suzuki cross-coupling reaction of 2, 5-dibromo-3-methylthiophene: Selectivity, characterization, DFT studies and their biological evaluations. Chem. Cent. J. 2018, 12, 49. [Google Scholar] [CrossRef]
- Nasr, T.; Bondock, S.; Eid, S. Design, synthesis, antimicrobial evaluation and molecular docking studies of some new thiophene, pyrazole and pyridone derivatives bearing sulfisoxazole moiety. Eur. J. Med. Chem. 2014, 84, 491–504. [Google Scholar] [CrossRef]
- Miao, Q.; Yan, X.; Zhao, K. Synthesis, Structure and Anticancer Activity Studies of 1-[(5-Bromo-2-thienyl) sulfonyl]-5-fluoro-1, 2, 3, 4-tetrahydropyrimidine-2, 4-dione. Chin. J. Chem. 2010, 28, 81–85. [Google Scholar] [CrossRef]
- Pulipati, L.; Sridevi, J.P.; Yogeeswari, P.; Sriram, D.; Kantevari, S. Synthesis and antitubercular evaluation of novel dibenzo [b, d] thiophene tethered imidazo [1, 2-a] pyridine-3-carboxamides. Bioorg. Med. Chem. Lett. 2016, 26, 3135–3140. [Google Scholar] [CrossRef]
- Deng, H.; Hu, J.; Hu, H.; He, M.; Fang, Y. Thieno [3, 2-b] thiophene-2-carboxylic acid derivatives as GPR35 agonists. Bioorg. Med. Chem. Lett. 2012, 22, 4148–4152. [Google Scholar] [CrossRef] [PubMed]
- Pandya, D.H.; Sharma, J.A.; Jalani, H.B.; Pandya, A.N.; Sudarsanam, V.; Kachler, S.; Klotz, K.N.; Vasu, K.K. Novel thiazole–thiophene conjugates as adenosine receptor antagonists: Synthesis, biological evaluation and docking studies. Bioorg. Med. Chem. Lett. 2015, 25, 1306–1309. [Google Scholar] [CrossRef] [PubMed]
- Baffoe, J.; Hoe, M.Y.; Toure, B.B. Copper-mediated N-heteroarylation of primary sulfonamides: Synthesis of mono-N-heteroaryl sulfonamides. Org. Lett. 2010, 12, 1532–1535. [Google Scholar] [CrossRef]
- Ali, E.H.; Nassar, F.I.; Badawi, A.; Afify, S.A. Physical properties and biological applications of novel substituted biphenyl-sulfonamides. Int. J. Genet. Mol. Biol. 2010, 2, 78–91. [Google Scholar]
- Perlovich, G.L.; Strakhova, N.N.; Kazachenko, V.P.; Volkova, T.V.; Tkachev, V.V.; Schaper, K.-J.; Raevsky, O.A. Sulfonamides as a subject to study molecular interactions in crystals and solutions: Sublimation, solubility, solvation, distribution and crystal structure. Int. J. Pharm. 2008, 349, 300–313. [Google Scholar] [CrossRef]
- Brown, G.M. The biosynthesis of folic acid. 2. Inhibition by sulfonamides. J. Biol. Chem. 1962, 237, 536–540. [Google Scholar] [CrossRef]
- Sherrill, C.D. Frontiers in electronic structure theory. J. Chem. Phys. 2010, 132, 110902. [Google Scholar] [CrossRef]
- Casely, I.J.; Ziller, J.W.; Fang, M.; Furche, F.; Evans, W.J. Facile Bismuth− Oxygen Bond Cleavage, C− H Activation, and Formation of a Monodentate Carbon-Bound Oxyaryl Dianion,(C6H2 t Bu2-3, 5-O-4) 2−. J. Am. Chem. Soc. 2011, 133, 5244–5247. [Google Scholar] [CrossRef] [PubMed]
- Dobson, J.F.; Vignale, G.; Das, M.P. Electronic Density Functional Theory: Recent Progress and New Directions; Springer Science & Business Media: Berlin, Germany, 2013. [Google Scholar]
- Ramachandran, K.; Deepa, G.; Namboori, K. Computational Chemistry and Molecular Modeling: Principles and Applications; Springer Science & Business Media: Berlin, Germany, 2008. [Google Scholar]
- Rasool, N.; Kanwal, A.; Rasheed, T.; Ain, Q.; Mahmood, T.; Ayub, K.; Zubair, M.; Khan, K.M.; Arshad, M.N.; M Asiri, A. One pot selective arylation of 2-bromo-5-chloro thiophene; molecular structure investigation via density functional theory (DFT), X-ray analysis, and their biological activities. Int. J. Mol. Sci. 2016, 17, 912. [Google Scholar] [CrossRef]
- Noreen, M.; Rasool, N.; Gull, Y.; Zubair, M.; Mahmood, T.; Ayub, K.; Nasim, F.-u.-H.; Yaqoob, A.; Zia-Ul-Haq, M.; De Feo, V. Synthesis, density functional theory (DFT), urease inhibition and antimicrobial activities of 5-aryl thiophenes bearing sulphonylacetamide moieties. Molecules 2015, 20, 19914–19928. [Google Scholar] [CrossRef] [PubMed]
- Noreen, M.; Rasool, N.; El Khatib, M.; Molander, G.A. Arylation and heteroarylation of thienylsulfonamides with organotrifluoroborates. J. Org. Chem. 2014, 79, 7243–7249. [Google Scholar] [CrossRef]
- Noreen, M.; Rasool, N.; Gull, Y.; Zahoor, A.F.; Yaqoob, A.; Kousar, S.; Zubair, M.; Bukhari, I.H.; Rana, U.A. A facile synthesis of new 5-aryl-thiophenes bearing sulfonamide moiety via Pd (0)-catalyzed Suzuki–Miyaura cross coupling reactions and 5-bromothiophene-2-acetamide: As potent urease inhibitor, antibacterial agent and hemolytically active compounds. J. Saudi Chem. Soc. 2017, 21, S403–S414. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision D. 01. 2010. Available online: https://researchcomputing.syr.edu/gaussian-09/ (accessed on 20 February 2021).
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Mansha, A.; Asim, S.; Zahoor, A.F.; Ghafoor, S.; Akbar, M.U. A computational perspective of vibrational and electronic analysis of potential photosensitizer 2-chlorothioxanthone. J. Mol. Struct. 2018, 1156, 571–582. [Google Scholar] [CrossRef]
- Stratmann, R.E.; Scuseria, G.E.; Frisch, M.J. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys. 1998, 109, 8218–8224. [Google Scholar] [CrossRef]
- Rad, A.S.; Ayub, K. Adsorption of thiophene on the surfaces of X12Y12 (X = Al, B, and Y = N, P) nanoclusters; A DFT study. J. Mol. Liq. 2017, 238, 303–309. [Google Scholar] [CrossRef]
- Mabkhot, Y.N.; Barakat, A.; Soliman, S.M.; El-Idreesy, T.T.; Ghabbour, H.A.; Al-Showiman, S.S. Synthesis, characterization and computational studies of a novel thieno [2, 3-b] thiophene derivative. J. Mol. Struct. 2017, 1130, 62–70. [Google Scholar] [CrossRef]
- Arshad, M.N.; Faidallah, H.M.; Asiri, A.M.; Kosar, N.; Mahmood, T. Structural, spectroscopic and nonlinear optical properties of sulfonamide derivatives; experimental and theoretical study. J. Mol. Struct. 2020, 1202, 127393. [Google Scholar] [CrossRef]
- Boufas, W.; Dupont, N.; Berredjem, M.; Berrezag, K.; Becheker, I.; Berredjem, H.; Aouf, N.-E. Synthesis and antibacterial activity of sulfonamides. SAR and DFT studies. J. Mol. Struct. 2014, 1074, 180–185. [Google Scholar] [CrossRef]
- Pandey, U.; Srivastava, M.; Singh, R.; Yadav, R. DFT study of conformational and vibrational characteristics of 2-(2-hydroxyphenyl) benzothiazole molecule. Spectrochim. Acta Part A 2014, 129, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Huang, X.R.; Chen, W.; Sun, C.C. Alkali metal atom-aromatic ring: A novel interaction mode realizes large first hyperpolarizabilities of M@ AR (M = Li, Na, and K, AR = pyrrole, indole, thiophene, and benzene). J. Comput. Chem. 2011, 32, 2005–2011. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.; Rasool, N.; Ikram, H.M.; Hashmi, M.A.; Mahmood, T.; Zubair, M.; Ahmad, G.; Rizwan, K.; Rashid, T.; Rashid, U. Synthesis of 3, 4-Biaryl-2, 5-Dichlorothiophene through Suzuki Cross-Coupling and Theoretical Exploration of Their Potential Applications as Nonlinear Optical Materials. Symmetry 2018, 10, 766. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Molecular Electrostatic Potentials; Taylor & Francis Group, LLC: New York, NY, USA, 2004. [Google Scholar]
- Belaidi, S.; Belaidi, H.; Bouzidi, D. Computational Methods Applied in Physical-Chemistry Property Relationships of Thiophene Derivatives. J. Comput. Theor. Nanosci. 2015, 12, 1737–1745. [Google Scholar] [CrossRef]
- Mathammal, R.; Sangeetha, K.; Sangeetha, M.; Mekala, R.; Gadheeja, S. Molecular structure, vibrational, UV, NMR, HOMO-LUMO, MEP, NLO, NBO analysis of 3, 5 di tert butyl 4 hydroxy benzoic acid. J. Mol. Struct. 2016, 1120, 1–14. [Google Scholar] [CrossRef]
- Ermiş, E. Synthesis, spectroscopic characterization and DFT calculations of novel Schiff base containing thiophene ring. J. Mol. Struct. 2018, 1156, 91–104. [Google Scholar] [CrossRef]
- Tanak, H.; Ağar, A.A.; Büyükgüngör, O. Experimental (XRD, FT-IR and UV–Vis) and theoretical modeling studies of Schiff base (E)-N′-((5-nitrothiophen-2-yl) methylene)-2-phenoxyaniline. Spectrochim. ActaPart A 2014, 118, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Mulya, F.; Santoso, G.A.; Aziz, H.A.; Pranowo, H.D. Design a better metalloporphyrin semiconductor: A theoretical studies on the effect of substituents and central ions. In Proceedings of the AIP Conference Proceedings, Melville, NY, USA, 21 July 2016; p. 080006. [Google Scholar]
- Djouambi, N.; Bougheloum, C.; Messalhi, A.; Bououdina, M.; Banerjee, A.; Chakraborty, S.; Ahuja, R. New concept on photocatalytic degradation of thiophene derivatives: Experimental and DFT studies. J. Phys. Chem. C 2018, 122, 15646–15651. [Google Scholar] [CrossRef]
- Tsuneda, T.; Song, J.-W.; Suzuki, S.; Hirao, K. On Koopmans’ theorem in density functional theory. J. Chem. Phys. 2010, 133, 174101. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, V.; Bhatt, A.; Dash, D.; Sharma, N. DFT calculations on molecular structures, HOMO–LUMO study, reactivity descriptors and spectral analyses of newly synthesized diorganotin (IV) 2-chloridophenylacetohydroxamate complexes. J. Comput. Chem. 2019, 40, 2354–2363. [Google Scholar] [CrossRef] [PubMed]
- Rauk, A. Orbital Interaction Theory of Organic Chemistry; John Wiley & Sons: Hoboken, NJ, UAS, 2004. [Google Scholar]
- Pearson, R.G. Chemical hardness and density functional theory. J. Chem. Sci. 2005, 117, 369–377. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).