
Expected and Unexpected Products in Half Curcuminoid Synthesis: Crystal Structures of But-3-en-2-ones and 3-Methylcyclohex-2-enones

 , ,
, ,  , and
, and
Abstract
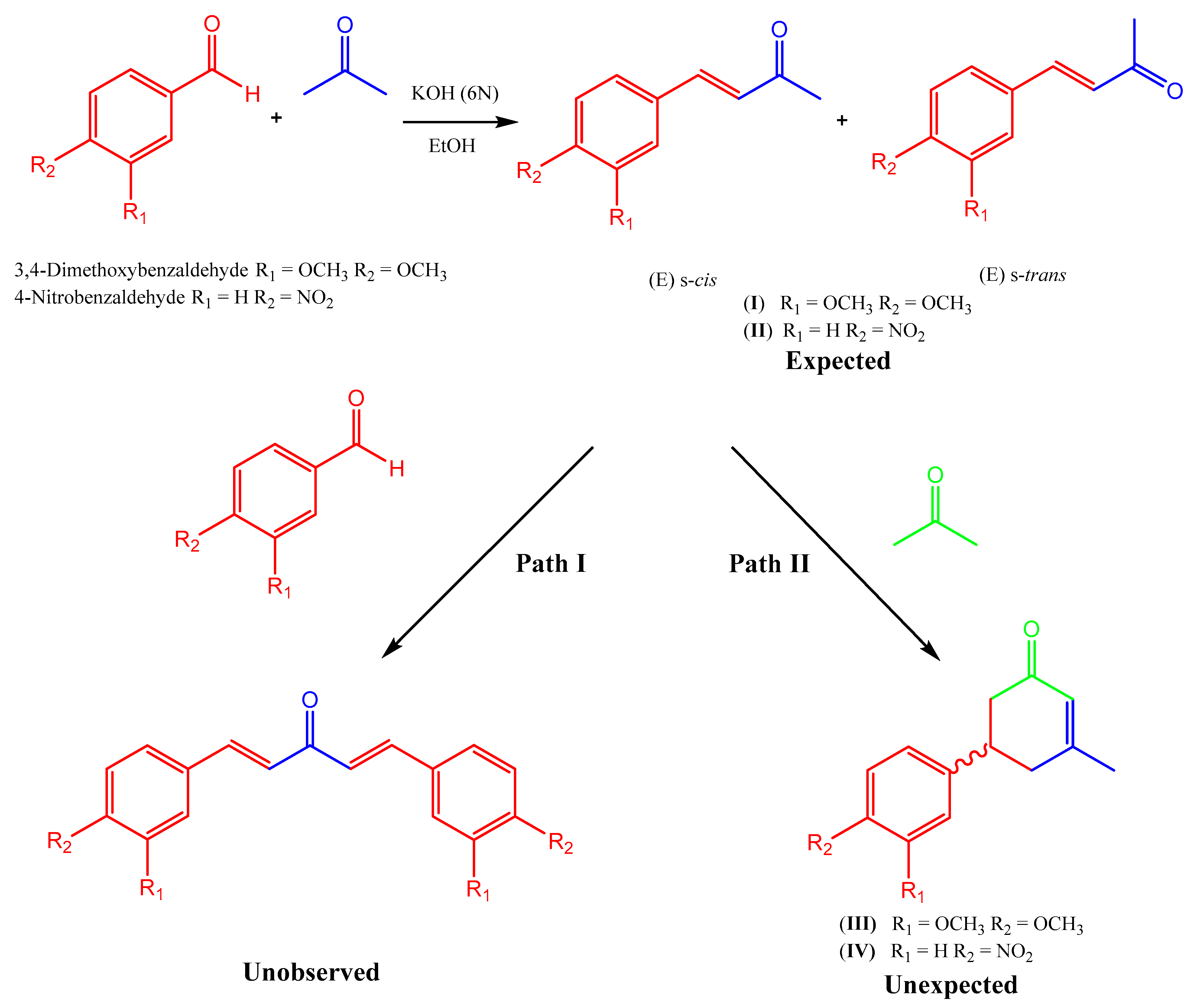
:1. Introduction
2. Materials and Methods
Synthesis and Crystallization
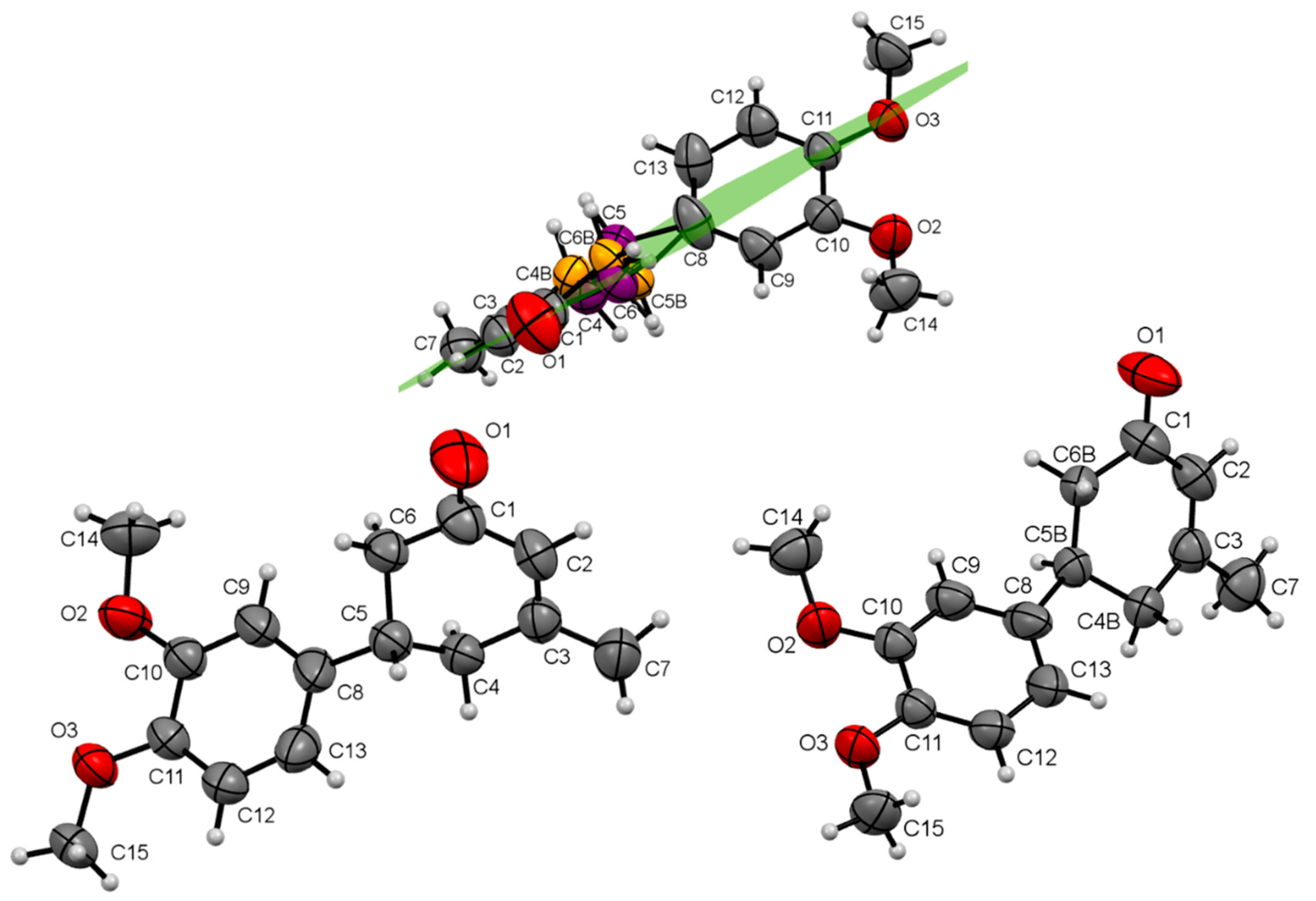
3. Results
4. Discussion
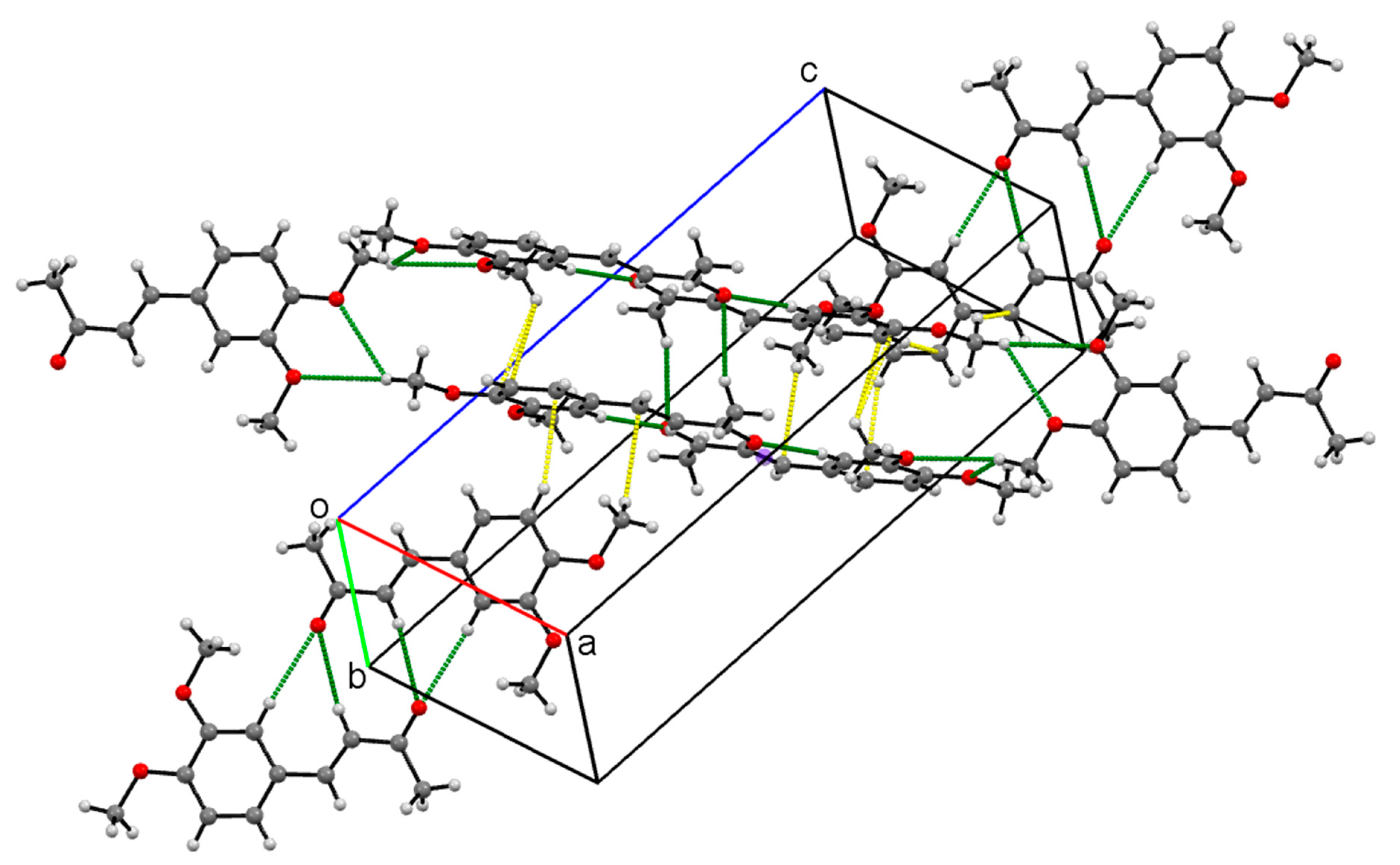
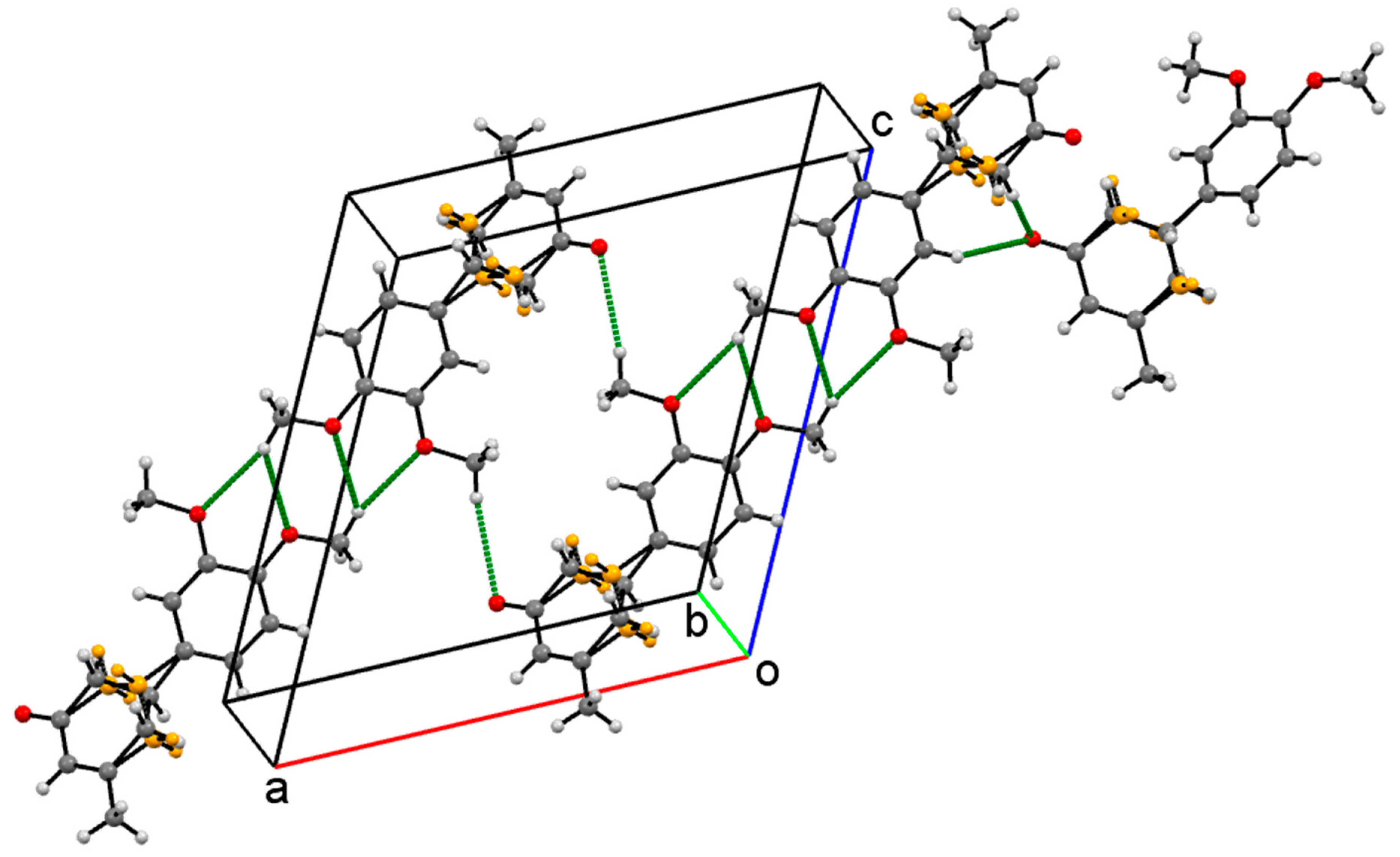
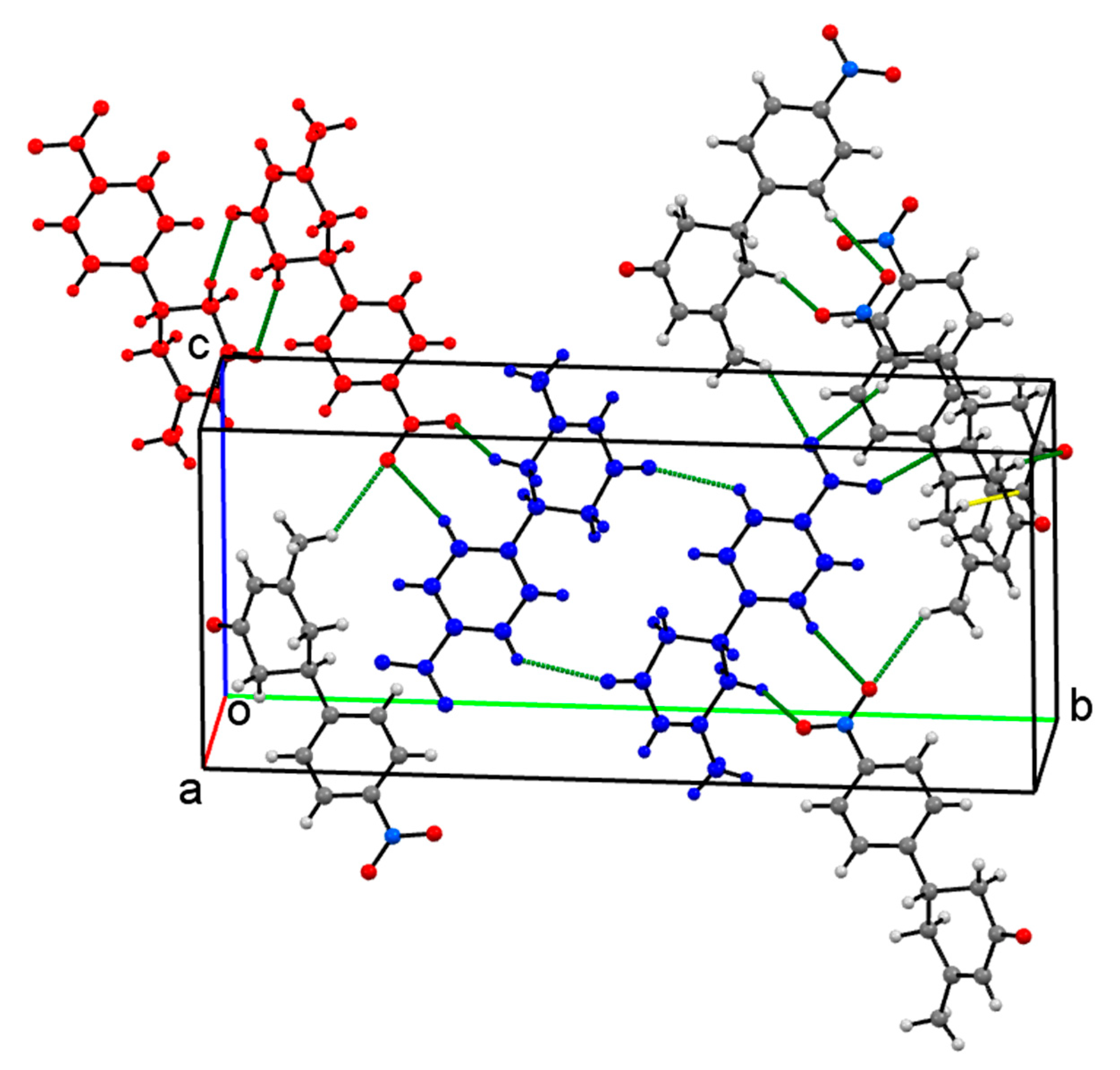
Crystal Packing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mapoung, S.; Mapoung, S.; Suzuki, S.; Fuji, S.; Naiki-Ito, A.; Kato, H.; Yodkeeree, S.; Yodkeeree, S.; Sakorn, N.; Sakorn, N.; et al. Dehydrozingerone, a Curcumin Analog, as a Potential Anti-Prostate Cancer Inhibitor in Vitro and in Vivo. Molecules 2020, 25, 2737. [Google Scholar] [CrossRef] [PubMed]
- Kondamudi, P.K.; Kovelamudi, H.; Nayak, P.G.; Rao, M.C.; Shenoy, R.R. Curcumin Half Analog Modulates Interleukin-6 and Tumor Necrosis Factor-Alpha in Inflammatory Bowel Disease. Pharmacogn. Mag. 2015, 11, S296–S302. [Google Scholar]
- Yogosawa, S.; Yamada, Y.; Yasuda, S.; Sun, Q.; Takizawa, K.; Sakai, T. Dehydrozingerone, a Structural Analogue of Curcumin, Induces Cell-Cycle Arrest at the G2/M Phase and Accumulates Intracellular ROS in HT-29 Human Colon Cancer Cells. J. Nat. Prod. 2012, 75, 2088–2093. [Google Scholar] [CrossRef] [PubMed]
- Suwito, H.; Jumina; Mustofa; Kristanti, A.N.; Puspaningsih, N.N.T. Chalcones: Synthesis, Structure Diversity and Pharmacological Aspects. J. Chem. Pharm. Res. 2014, 6, 1076–1088. [Google Scholar]
- Mousavi, M.R.; Maghsoodlou, M.T.; Gharari, H. Sodium Carbonate-Catalyzed Claisen-Schmidt Condensation: One-Pot Synthesis of Highly Functionalized Cyclohexenones under Environmental Conditions. Res. Chem. Intermed. 2016, 42, 2233–2246. [Google Scholar] [CrossRef]
- Rahman, A.F.M.M.; Ali, R.; Jahng, Y.; Kadi, A.A.A. Facile Solvent Free Claisen-Schmidt Reaction: Synthesis of α,α’-bis-(Substituted-Benzylidene)cycloalkanones and α,α’-bis-(Substituted-Alkylidene)cycloalkanones. Molecules 2012, 17, 571–583. [Google Scholar] [CrossRef] [Green Version]
- Sreevidya, T.V.; Narayana, B.; Yathirajan, H.S. Synthesis and Characterization of Some Chalcones and Their Cyclohexenone Derivatives. Cent. Eur. J. Chem. 2010, 8, 174–181. [Google Scholar] [CrossRef]
- Gallier, F.; Martel, A.; Dujardin, G. Enantioselective Access to Robinson Annulation Products and Michael Adducts as Precursors. Angew. Chemie Int. Ed. 2017, 56, 12424–12458. [Google Scholar] [CrossRef]
- Ghorai, M.K.; Samanta, S.; Das, S. Synthesis of 3,5-Disubstituted Cyclohex-2-en-1-one via a Five-Step Domino Reaction Catalyzed by Secondary Amines: Formation of (e)-α,β-Unsaturated Methyl Ketones. Asian J. Org. Chem. 2013, 2, 1026–1030. [Google Scholar] [CrossRef]
- Xiang, Z.; Liang, Y.; Chen, X.; Wu, Q.; Lin, X. D-Aminoacylase-Initiated Cascade Aldol Condensation/Robinson Annulation for Synthesis of Substituted Cyclohex-2-enones from Simple Aldehydes and Acetone. Amino Acids 2014, 46, 1929–1937. [Google Scholar] [CrossRef]
- Jasinski, J.P.; Golen, J.A.; Samshuddin, S.; Narayana, B.; Yathirajan, H.S. Ethyl 2-amino-4,6-bis-(4-fluorophenyl)cyclohexa-1,3-diene-1-carboxylate. Acta Crystallogr. Sect. E Struct. Reports Online 2012, 68, o585. [Google Scholar] [CrossRef] [PubMed]
- Rai, S.; Patel, P.N.; Chadha, A. Preparation, Characterisation, and Crystal Structure Analysis of (2E,2′E)-3,3′-(1,4-phenylene)bis(1-(2-aminophenyl)prop-2-en-1-one. Crystallogr. Reports 2016, 61, 1086–1089. [Google Scholar] [CrossRef]
- Singh, V.D.; Salian, V.V.; Narayana, B.; Sarojini, B.K.; Kamni; Anthal, S.; Kant, R. Synthesis and Crystal Structure of a Chalcone Derivative. Crystallogr. Reports 2017, 62, 1157–1159. [Google Scholar] [CrossRef]
- Wang, F.; Liu, Y.; Qi, Z.; Dai, W.; Li, X. Rhodium-Catalyzed Tandem Aldol Condensation-Robinson Annulation Between Aldehydes and Acetone: Synthesis of 3-methylcyclohexenones. Tetrahedron Lett. 2014, 55, 6399–6402. [Google Scholar] [CrossRef]
- MNova Software. Available online: https://mestrelab.com/download/mnova/ (accessed on 10 April 2021).
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Van De Streek, J.; Wood, P.A. Mercury CSD 2.0-New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Collins, A.; Barr, G.; Dong, W.; Gilmore, C.J.; Middlemiss, D.S.; Parkin, A.; Wilson, C.C. The Application of Cluster Analysis to Identify Conformational Preferences in Enones and Enimines from Crystal Structural Data. Acta Crystallogr. Sect. B Struct. Sci. 2007, 63, 469–476. [Google Scholar] [CrossRef]
- Wang, Y.H.; Zou, J.W.; Zhang, B.; Lu, Y.X.; Jin, H.X.; Yu, Q. Sen Enone-Dienol Tautomerism of But-2-enal and Substituent Effect: A Theoretical Study. J. Mol. Struct. THEOCHEM 2005, 755, 31–37. [Google Scholar] [CrossRef]
- Zhang, H.; Li, S.; Shi, X. (E)-2-Methoxy-4-(3-oxobut-1-enyl)phenyl Acetate. Acta Crystallogr. Sect. E Struct. Rep. Online 2008, 64, o1507. [Google Scholar] [CrossRef] [PubMed]
- Patai, S.; Rappopor, Z. The Chemistry of Enones Part 1; Wiley: Hoboken, NJ, USA, 1989; pp. 1–1261. [Google Scholar]
- Lin, F.F.S.; Servis, K.L. Nuclear Magnetic Resonance Spectroscopy. Rotational Isomerism in α,β-Unsaturated Acyl Fluorides. J. Am. Chem. Soc. 1972, 94, 5794–5801. [Google Scholar] [CrossRef]
- Loncharich, R.J.; Schwartz, T.R.; Houk, K.N. Theoretical studies of conformations of acrolein, acrylic acid, methyl acrylate, and their Lewis acid complexes. J. Am. Chem. Soc. 1987, 109, 14–23. [Google Scholar] [CrossRef]
- Cremer, D.; Pople, J.A. A General Definition of Ring Puckering Coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Chion, B.; Lajzerowicz, J.; Bordeaux, D.; Collet, A.; Jacques, J. Structural aspects of solid solutions of enantiomers. The 3-hydroxymethyl- and 3-carboxy-2,2,5,5-tetramethylpyrrolidinyl 1-oxyl systems as examples. J. Phys. Chem. 1978, 82, 2682–2688. [Google Scholar] [CrossRef]
- Rekis, T.; Bērziņš, A. On the structural aspects of solid solutions of enantiomers: An intriguing case study of enantiomer recognition in the solid state. CrystEngComm 2018, 20, 6909–6918. [Google Scholar] [CrossRef]
- Brandel, C.; Petit, S.; Coquerel, Y.C. Structural Aspects of Solid Solutions of Enantiomers. Curr. Pharm. Des. 2016, 22, 4929–4941. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Gupta, M. A simple and efficient method for selective single aldol condensation between arylaldehydes and acetone. Synth. Commun. 2005, 35, 213–222. [Google Scholar] [CrossRef]
- Bounora, P.T.; Rosauer, K.G.; Dai, L. Control of the Aqueus Aldol Addition Under Claisen-Schmidt Condition. Tetrahedron Lett. 1995, 36, 4009–4012. [Google Scholar] [CrossRef]
- Bethi, V.; Fernandes, R.A. Traceless OH-Directed Wacker Oxidation-Elimination, an Alternative to Wittig Olefination/Aldol Condensation: One-Pot Synthesis of α,β-Unsaturated and Nonconjugated Ketones from Homoallyl Alcohols. J. Org. Chem. 2016, 81, 8577–8584. [Google Scholar] [CrossRef]
- Walker, G.N. Triton B in Synthesis of 3-Phenylcyclohexenones. J. Am. Chem. Soc. 1955, 77, 3664–3667. [Google Scholar] [CrossRef]
- Surya Prakash Rao, H.; Jothilingam, S. Solvent-free microwave-mediated Michael addition reactions. J. Chem. Sci. 2005, 117, 323–328. [Google Scholar]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | I | II | III | IV |
|---|---|---|---|---|
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Unit cell parameters [Å,°] | a = 9.6000(7) b = 5.3426(4) c = 22.353(2) β = 97.307(2) | a = 7.4561(4) b = 7.2591(4) c = 17.7744(10) β = 94.505(2) | a = 13.0205(10) b = 8.0494(6) c = 14.1040(11) β = 114.852(2) | a = 4.9301(18) b = 23.652(9) c = 10.014(4) β = 97.692(12) |
| Volume [Å3] | 1137.17(14) | 959.06(9) | 1341.32(18) | 1157.2(8) |
| Z/Calculated density [mg/m3] | 4/1.205 | 4/1.324 | 4/1.220 | 4/1.327 |
| Z′ | 1 | 1 | 1 | 1 |
| Absorption coefficient [mm−1] | 0.086 | 0.099 | 0.084 | 0.095 |
| Space group, F (000) | P21/n, 440 | P21/n, 400 | P21/c, 528 | P21/c, 488 |
| Index ranges | −11 ≤ h ≤ 11 −6 ≤ k ≤ 6 −25 ≤ l ≤ 26 | −8 ≤ h ≤ 9 −9 ≤ k ≤ 9 −23 ≤ l ≤ 21 | −15 ≤ h ≤ 15 −8 ≤ k ≤ 9 −11 ≤ l ≤ 16 | −6 ≤ h ≤ 6 −30 ≤ k ≤ 30 −13 ≤ l ≤ 13 |
| Reflections collected | 6066 | 16,684 | 5906 | 31,441 |
| Independent reflections | 2071 [R(int) = 0.0722] | 2446 [R(int) = 0.0940] | 2442 [R(int) = 0.1320] | 2663 [R(int) = 0.1187] |
| Observed reflections (I ≥ 2σ(I)) | 1100 | 935 | 1383 | 1290 |
| Completeness to θ = 25.10 | 99.6% | 99.8% | 99.6% | 99.7% |
| Data/restraints/parameters | 2071/0/139 | 2194/247/441 | 2442/ 72/195 | 2663/0/155 |
| Final R indices (I ≥ 2σ(I)) | R1 = 0.0482 wR2 = 0.1026 | R1 = 0.0532 wR2 = 0.1047 | R1 = 0.0631 wR2 = 0.1370 | R1 = 0.0516 wR2 = 0.0907 |
| R indices (all data) | R1 = 0.1005 wR2 = 0.1304 | R1 = 0.1416 wR2 = 0.1670 | R1 = 0.1204 wR2 = 0.1712 | R1 = 0.1469 wR2 = 0.1203 |
| Goodness-of-fit on F2 | 0.940 | 0.995 | 1.025 | 1.005 |
| Largest diff. peak/hole [e·Å−3] | 0.197/−0.172 | 0.114/−0.131 | 0.198/−0.268 | 0.174/−0.139 |
| CCDC deposit number | 2075964 | 2075965 | 2075966 | 2075970 |
| Compound | D-H···A | D-H | H···A | D···A | D-H···A | Symmetry Code |
|---|---|---|---|---|---|---|
| I | C1–H1A···O1 C3–H3···O1 C6–H6···O1 C11–H11B···Cg1 C9–H9···Cg1 C12–H12C···C3,4 | 0.960 0.930 0.930 0.960 0.930 0.960 | 2.61 2.60 2.62 2.84 3.05 2.84 | 3.529(3) 3.503(2) 3.545(3) 3.724(2) 3.733(2) 3.751(2) | 160.3 162.4 173.8 154.0 131.9 143.3 | x, −1+y, z 1−x, 1−y, 1−z 1−x, 1−y, 1−z x, 1+y, z 1/2−x, −1/2+y, 1/2−z 1/2−x, −1/2+y, 1/2−z |
| II | C7–H7···O2 C9–H9···O3 C4-H4···O1 C10–H10···O1 | 0.930 0.930 0.930 0.930 | 2.57 2.57 2.72 2.72 | 3.327(9) 3.301(9) 3.559(7) 3.539(6) | 138.9 135.8 150.0 147.9 | −x, −y, 1−z −x+1/2, y+1/2, −z+3/2 1−x, 2−y, 1−z 1−x, 2−y, 1−z |
| III | C6–H6A···O1 C9–H9···O1 C15–H15A··· O2 C15–H15A···O3 C14–H14A···O1 C4B–H4C···O3 | 0.970 0.930 0.960 0.960 0.960 0.970 | 2.59 2.65 2.62 2.80 2.86 2.66 | 3.539(10) 3.467(3) 3.519(3) 3.446(3) 3.806(3) 3.585(10) | 164.5 146.3 155.5 126.0 170.0 159.0 | −x, −y+1, −z+1 1−x, y+1/2, −z+1/2 −x, 1−y, 1−z −x, 1−y, 1−z 1−x, 1−y, 1−z x, 1/2−y, −1/2+z |
| IV | C6–H6A···O1 C6–H6B···O1 C7–H7B···O3 C12–H12···O1 C4–H4A···O2 C9–H9···O3 C5–H5···Cg1 C4–H4B···C2,3 | 0.970 0.970 0.960 0.930 0.970 0.930 0.980 0.970 | 2.64 2.88 2.68 2.66 2.72 2.73 2.98 2.84 | 3.581(3) 3.829(3) 3.467(3) 3.343(3) 3.327(3) 3.630(3) 3.824(3) 3.788(3) | 162.7 166.0 139.0 131.0 121.0 162.0 145.0 165.0 | −1+x, y, z −x, 1−y, 1−z −1+x, y,1+z 1−x,1−y, 1−z −1+x, 1/2−y, 1/2+z −1+x, 1/2−y, 1/2+z −1+x, y, z −1+x, y, z |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Obregón-Mendoza, M.A.; Arias-Olguín, I.I.; Meza-Morales, W.; Alvarez-Ricardo, Y.; Chávez, M.I.; Toscano, R.A.; Cassani, J.; Enríquez, R.G. Expected and Unexpected Products in Half Curcuminoid Synthesis: Crystal Structures of But-3-en-2-ones and 3-Methylcyclohex-2-enones. Crystals 2021, 11, 404. https://doi.org/10.3390/cryst11040404
Obregón-Mendoza MA, Arias-Olguín II, Meza-Morales W, Alvarez-Ricardo Y, Chávez MI, Toscano RA, Cassani J, Enríquez RG. Expected and Unexpected Products in Half Curcuminoid Synthesis: Crystal Structures of But-3-en-2-ones and 3-Methylcyclohex-2-enones. Crystals. 2021; 11(4):404. https://doi.org/10.3390/cryst11040404
Chicago/Turabian StyleObregón-Mendoza, Marco A., Imilla I. Arias-Olguín, William Meza-Morales, Yair Alvarez-Ricardo, María Isabel Chávez, Rubén A. Toscano, Julia Cassani, and Raúl G. Enríquez. 2021. "Expected and Unexpected Products in Half Curcuminoid Synthesis: Crystal Structures of But-3-en-2-ones and 3-Methylcyclohex-2-enones" Crystals 11, no. 4: 404. https://doi.org/10.3390/cryst11040404