With the excellent arc erosion resistance, abrasion resistance, switch operation characteristics and good resistance to welding, AgSnO
2 has become a promising material to replace AgCdO electrical contact materials [
1]. However, AgSnO
2 contact material has many shortcomings such as large contact resistance, high temperature rise, and processing difficulties, which limit its use. SnO
2 is a wide-bandgap semiconductor material, so it increases the resistance and temperature rise of contacts and has a great impact on the thermal conductivity of contacts [
2]. What is more, SnO
2 has a high hardness, leading to the poor plasticity and ductility of AgSnO
2 contact materials and the difficulties of contact modeling [
3].
Improving the performance of AgSnO
2 contact materials by doping has always been an important research direction. Reference [
4] used powder metallurgy to prepare Bi-doped AgSnO
2 contact materials and analyzed the influence of Bi-doping on electrical properties. Reference [
5] used the internal oxidation method to prepare different concentrations of NiO doped AgSnO
2 contact materials, and analyzed the influence of different concentrations of doping on the mechanical properties. The experimental results of references [
4,
5] show that doping can improve the electrical and mechanical properties of AgSnO
2 contact materials. However, in the use of experimental methods to improve the electrical and mechanical properties of AgSnO
2 contact materials, the types and proportions of doping elements rely on "trial and error" methods, which will cause a lot of waste of financial and material resources. Therefore, the current research on doping to improve the electrical and mechanical properties of AgSnO
2 contact materials urgently needs effective theoretical support. SnO
2, as the reinforcing phase in the AgSnO
2 contact material, directly affects the performance of the AgSnO
2 contact material. Therefore, the performance of the AgSnO
2 contact material can be improved by improving the performance of SnO
2. Doping non-metallic element N can reduce the band gap of SnO
2 and increase the conductivity of SnO
2. Moreover, since the ionic radii of N and O are the closest, it is easier for N atoms to replace O atoms than other non-metallic elements [
6]. According to the co-doping theory proposed by Yamamoto and Katayama, the co-doping of two or more elements can further improve the material properties [
7]. In this study, the conductivity and toughness of SnO
2 are improved by doping, and the wettability between SnO
2 and Ag is also improved, which can prevent SnO
2 from precipitating out of the Ag liquid to form an enriched area, and reduce the contact resistance of AgSnO
2. Compared with the traditional powder metallurgy process, the SnO
2 particles prepared by the sol-gel method are smaller and can be evenly distributed in the Ag matrix, which can improve the processing performance of the AgSnO
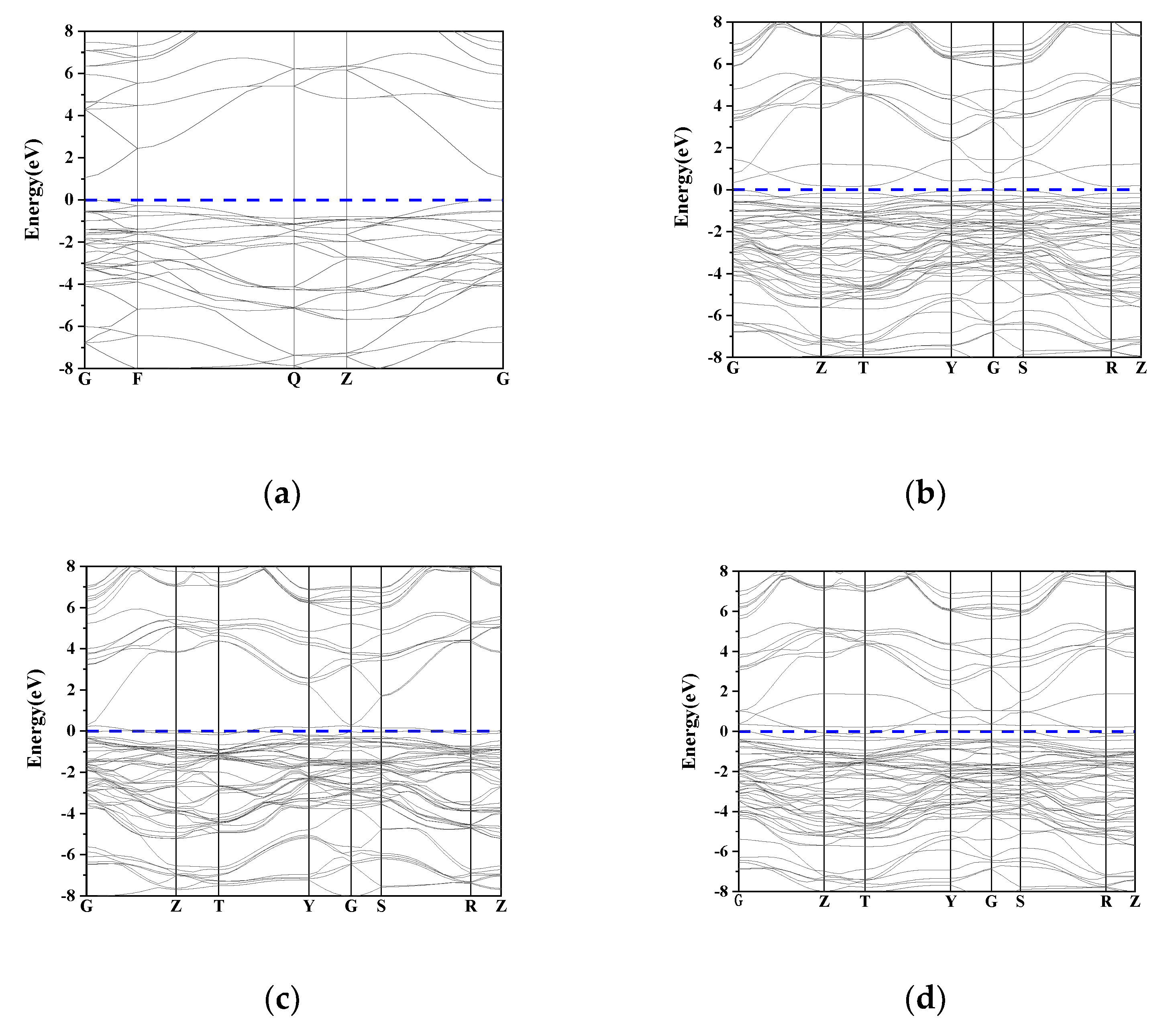
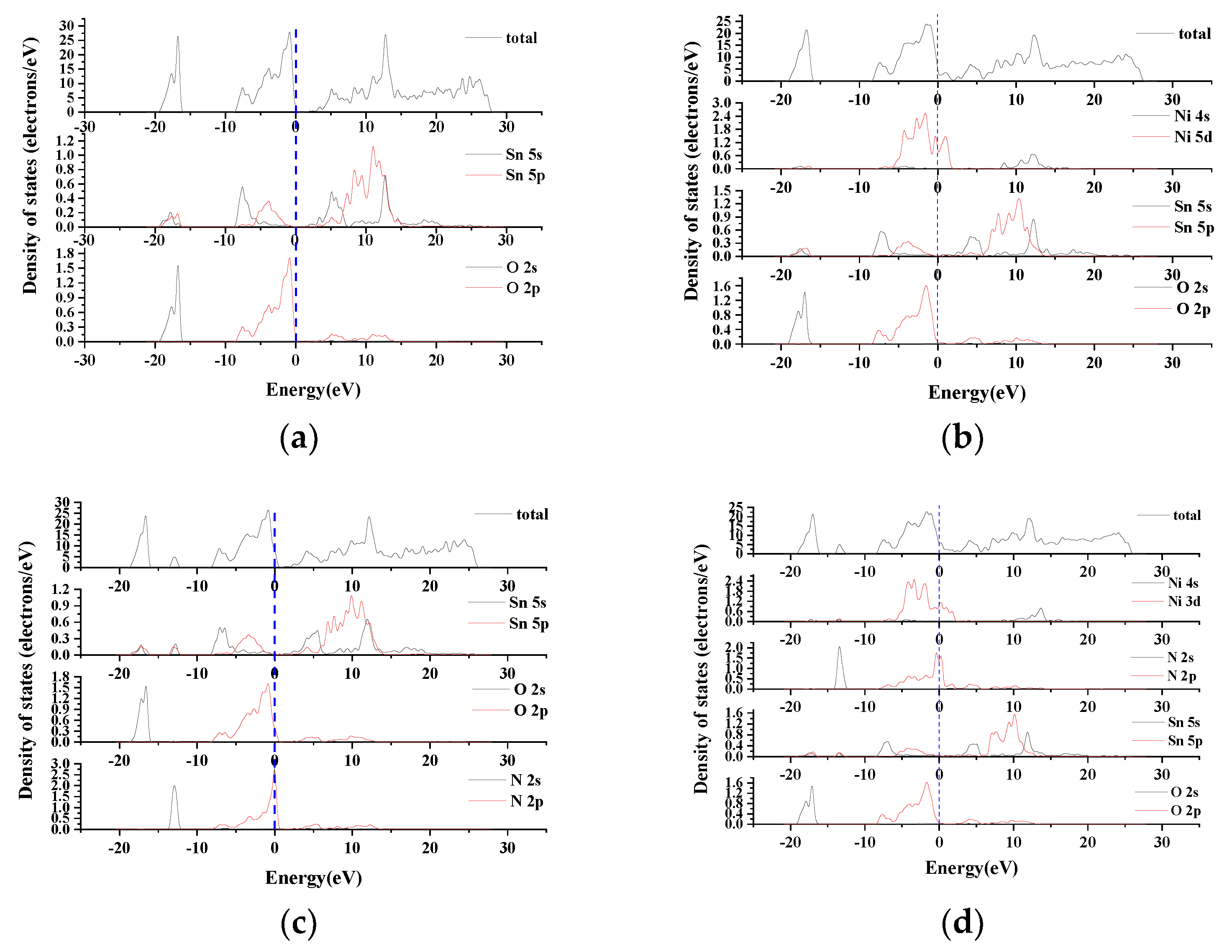
2 contact material. In this paper, the first principles based on density functional theory are used to calculate the band structure, density of states, layout elastic modulus and thermodynamic parameters of pure SnO
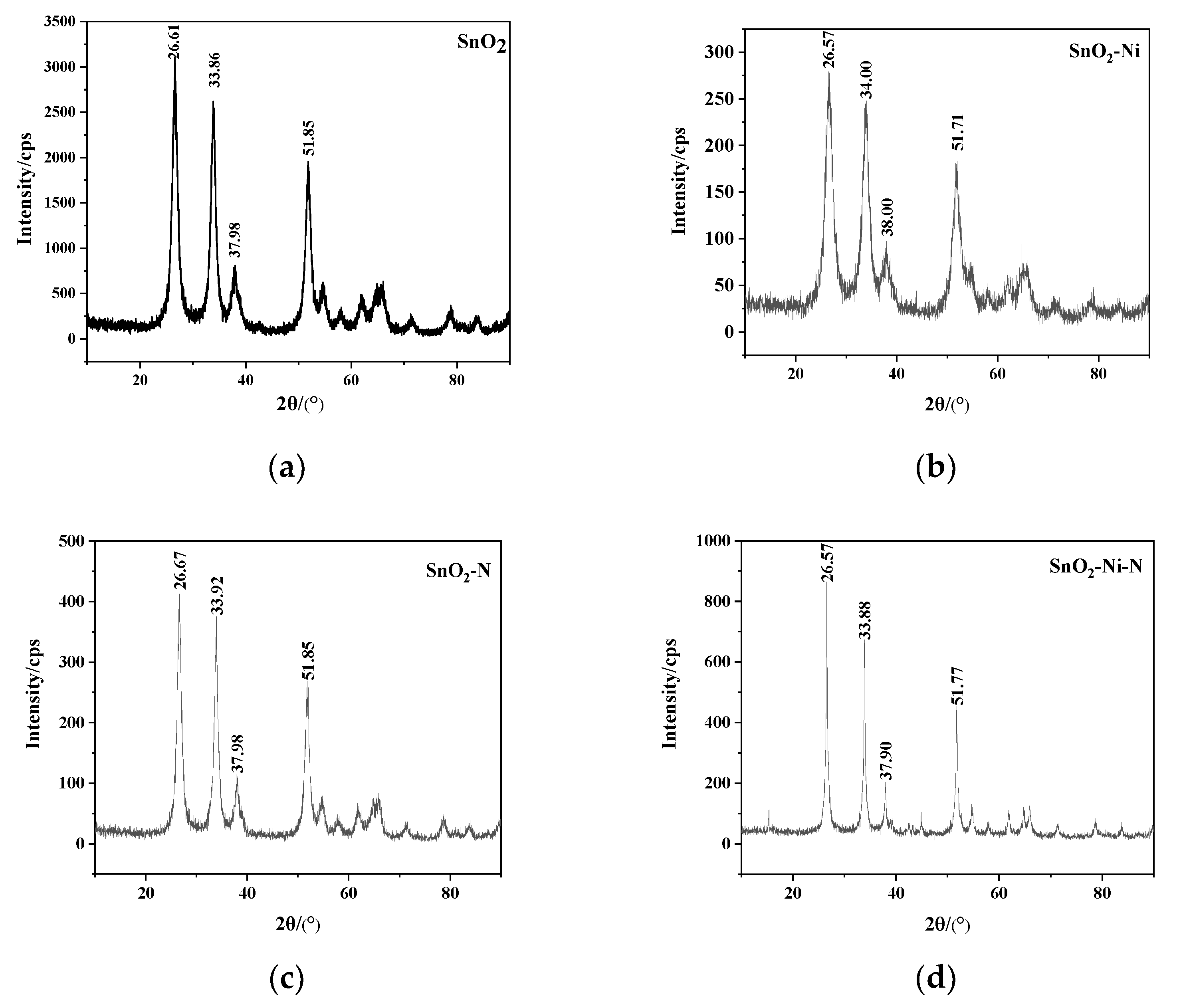
2, single doped elements Ni, N, and co-doped Ni and N. To analyze the influence of conductivity and mechanical properties before and after doping, the sol-gel method was used to prepare SnO
2 powder with different additives, and the influence of different additives on the wetting angle of Ag and SnO
2 was measured. Then, the powder metallurgy method was used to prepare the AgSnO
2 contact material. Hardness, electrical conductivity and contact resistance are tested to analyze their performance, providing a reference for the further development of AgSnO
2 contact materials.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}