
Molecular Dynamics Simulation of β-HMX Crystal Morphology Induced by Polymer Additives


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Methods
2.2. Simulation Details
3. Results and Discussion
3.1. Crystal Morphology of β-HMX in a Vacuum
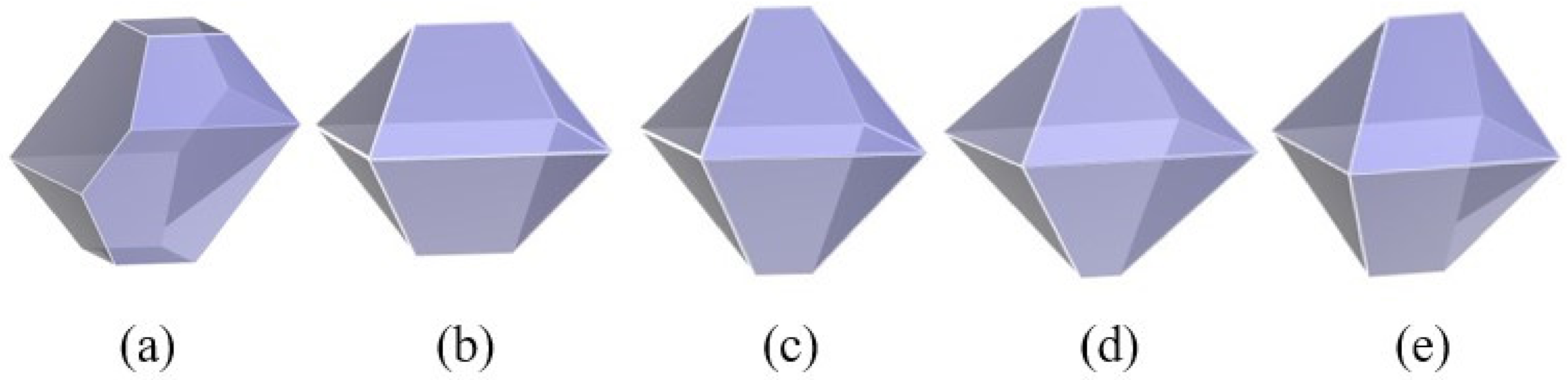
3.2. Effect of the Water Ratio on the Morphology of β-HMX
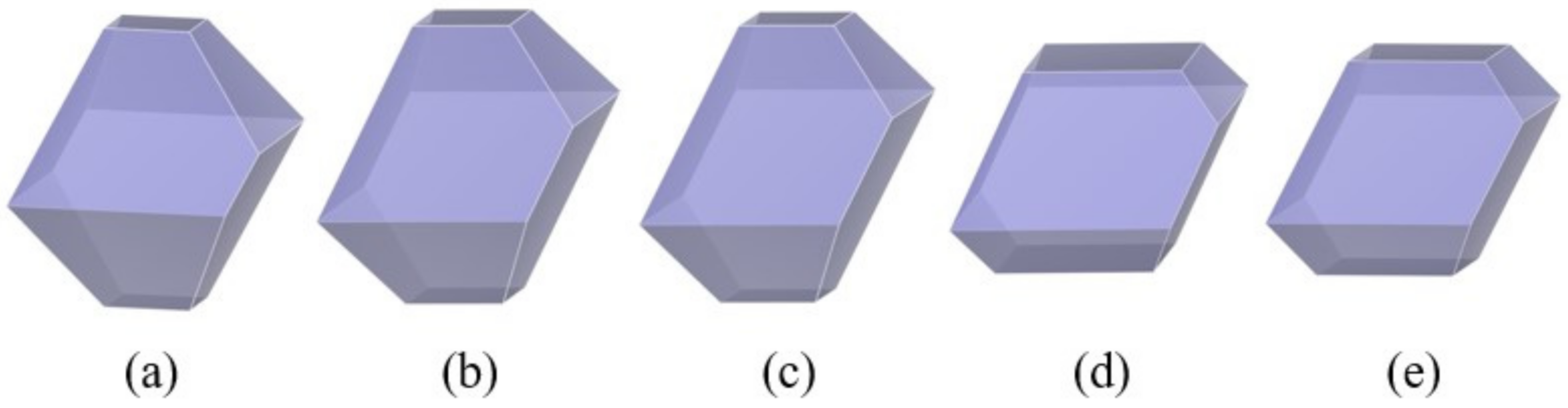
3.3. Effect of the Chain Length of Polymer Additives on the Morphology of β-HMX
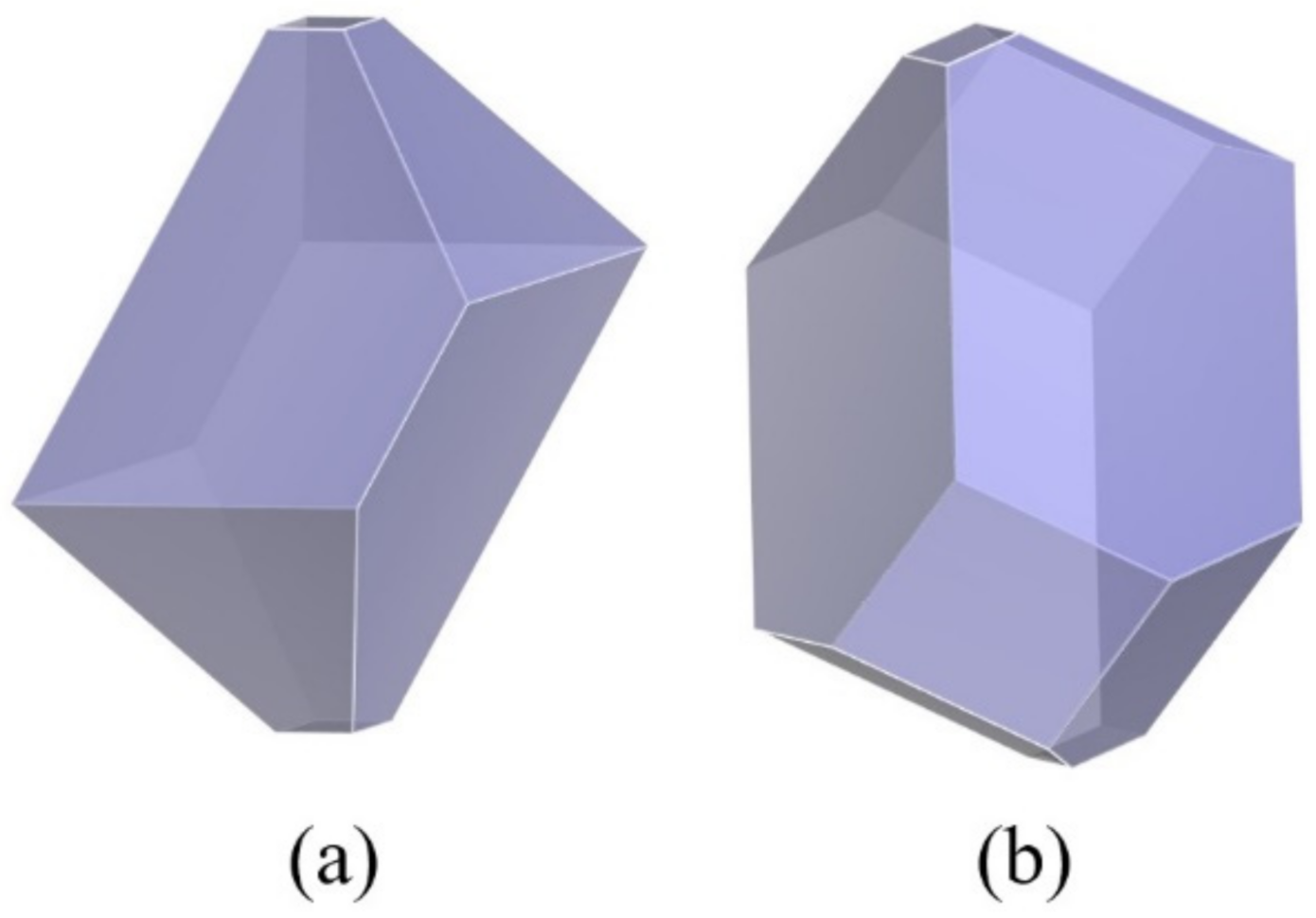
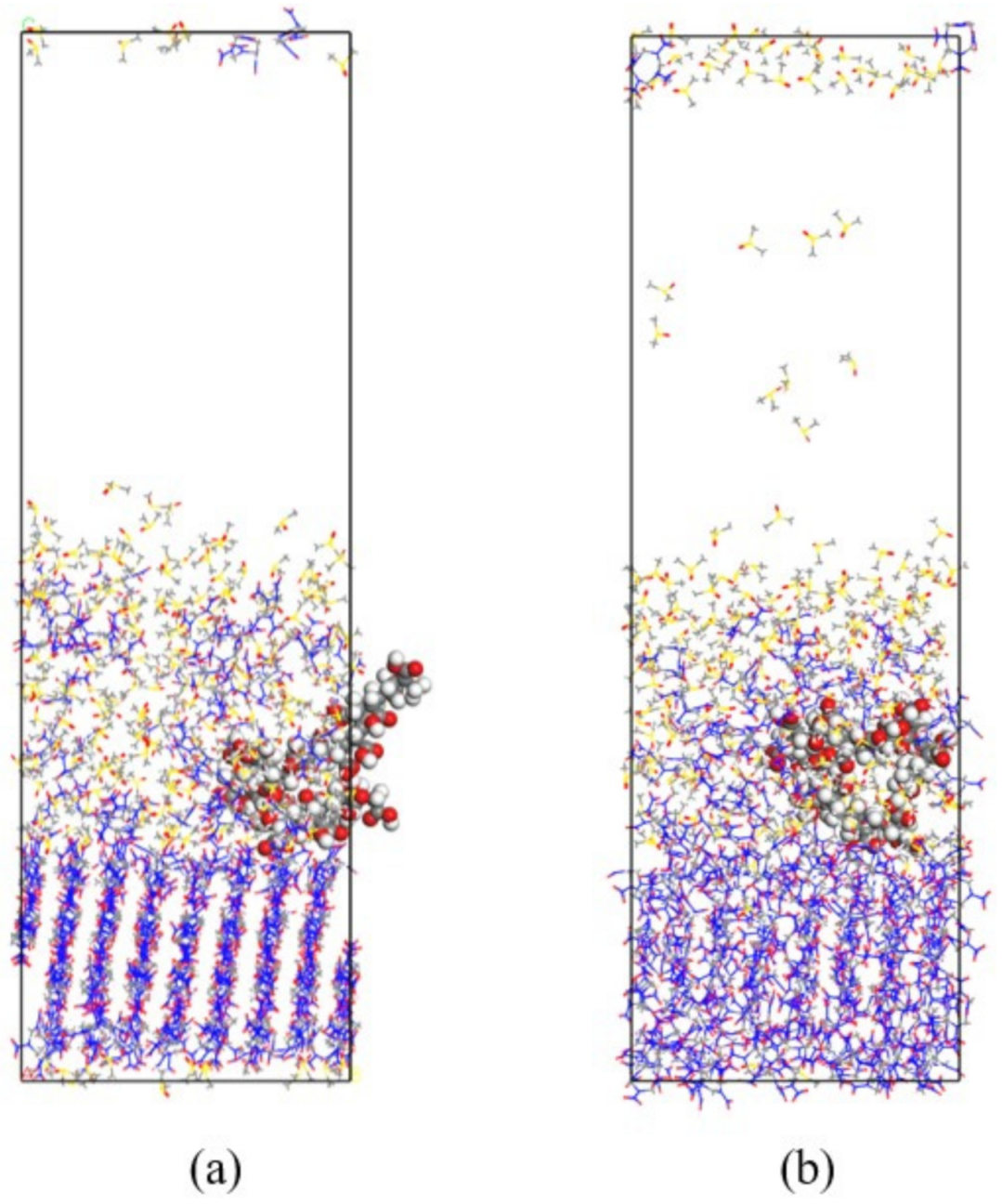
3.4. Effect of Different Polymer Additives on the Morphology of β-HMX Crystal
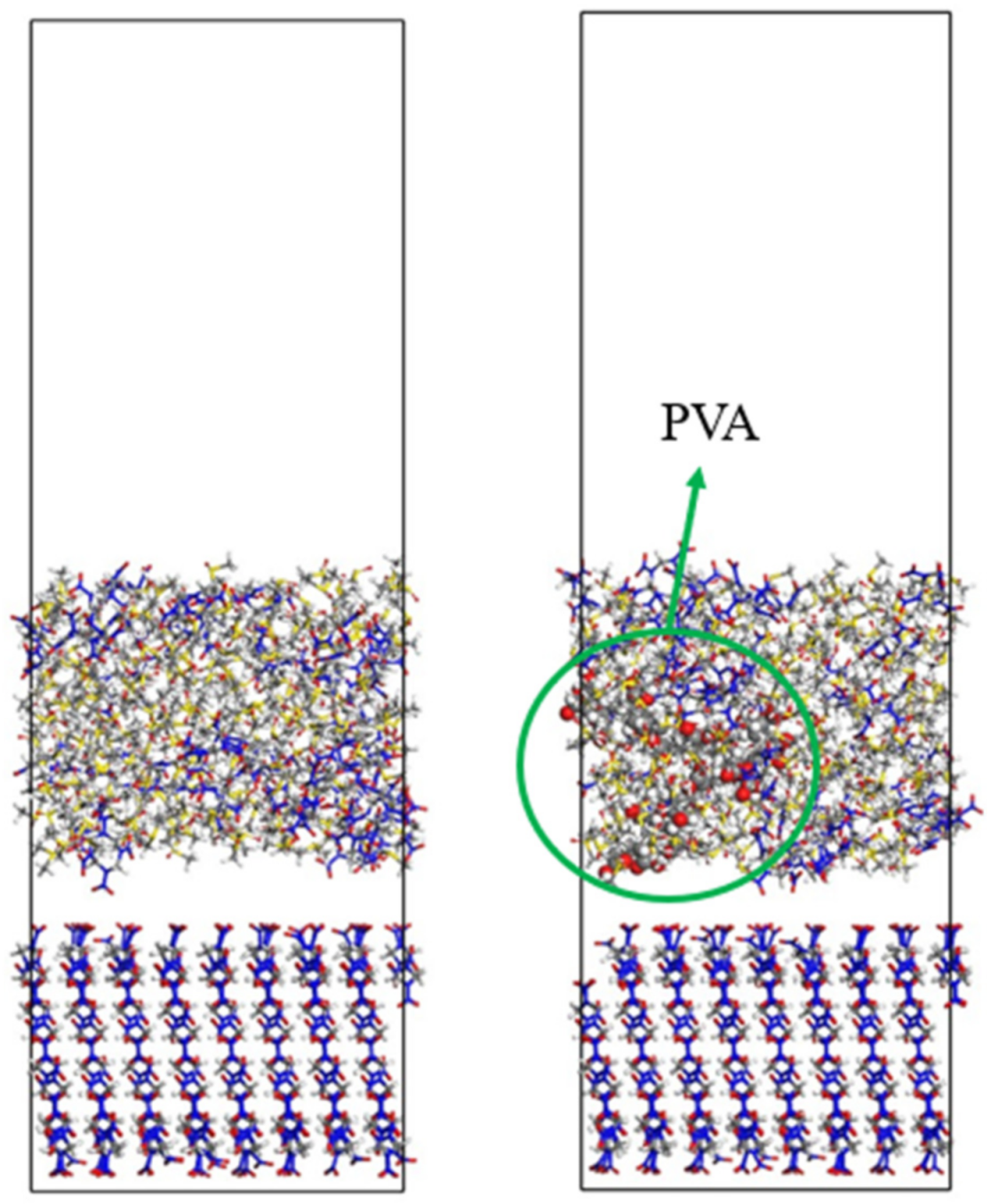
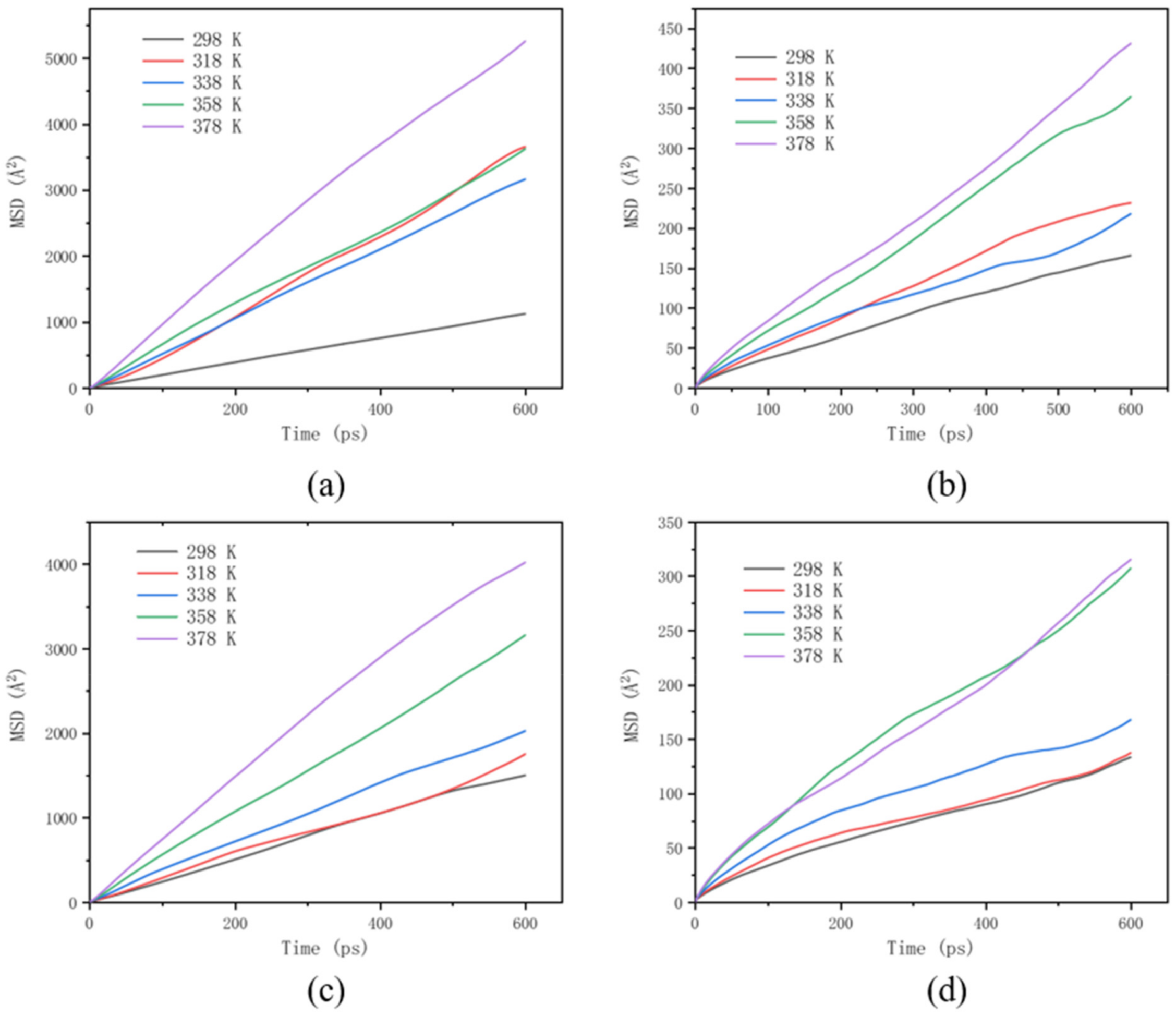
3.5. Effect of Polymer Additives on Molecular Motion Ability in Solution
4. Conclusions
- (1)
- The crystal morphology of β-HMX in a vacuum obtained by the attachment energy model shows that the areas of the main crystal planes were {0 1 1} > {1 1 −1} > {0 2 0} > {1 0 0} > {1 0 −2}. The {1 0 0} plane had the largest polarity and uneven surface because the nitro groups were perpendicular to the crystal surface and the structure was loose with voids.
- (2)
- The simulation results of the β-HMX crystal morphology in the presence of water were compared with the experimental results, indicating that a small amount of water affected the size of the {1 0 0} plane, and the {1 0 0} plane disappeared completely in the presence of a large amount of water.
- (3)
- The interaction of the solvent box composed of solvent DMSO and polymer PVA with different chain lengths on different crystal faces of β-HMX was studied, and the crystal morphology was predicted. The results show that the relative molecular mass of the polymer additive was 5% of the solvent mass, and the modification effect of the additive on the crystal morphology could be observed in the simulation calculation.
- (4)
- The crystal morphology of β-HMX in the presence of two additives was simulated and predicted, and the molecular dynamics characteristics of solute and solvent molecules in the crystallization process were studied. The results show that PVA blocked the movement of solvent and solute molecules, which may have affected the transport process and even the crystal morphology.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Naya, T.; Kohga, M. Influences of particle size and content of HMX on burning characteristics of HMX-based propellant. Aerosp. Sci. Technol. 2013, 27, 209–215. [Google Scholar] [CrossRef]
- Samudre, S.S.; Nair, U.R.; Gore, G.M.; Sinha, R.K.; Sikder, A.K.; Asthana, S.N. Studies on an Improved Plastic Bonded Explosive (PBX) for Shaped Charges. Propellants Explos. Pyrotech. 2009, 34, 145–150. [Google Scholar] [CrossRef]
- Vadhe, P.P.; Pawar, R.B.; Sinha, R.K.; Asthana, S.N.; Rao, A.S. Cast aluminized explosives (review). Combust. Explos. Shock Waves 2008, 44, 461–477. [Google Scholar] [CrossRef]
- Pereverzev, A.; Sewell, T. Elastic Coefficients of β-HMX as Functions of Pressure and Temperature from Molecular Dynamics. Cryst. 2020, 10, 1123. [Google Scholar] [CrossRef]
- Burch, A.C.; Yeager, J.D.; Bahr, D.F. Nanoindentation of HMX and Idoxuridine to Determine Mechanical Similarity. Crystals 2017, 7, 335. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.-X.; Chen, S.-S.; Li, Y.-Y.; Yang, J.-Y.; Wei, T.-Y.; Jin, S.-H. An Investigation into the Effects of Additives on Crystal Characteristics and Impact Sensitivity of RDX. J. Energetic Mater. 2013, 32, 184–198. [Google Scholar] [CrossRef]
- Rane, C.V.; Ekambara, K.; Joshi, J.B.; Ramkrishna, D. Effect of impeller design and power consumption on crystal size distribution. AIChE J. 2014, 60, 3596–3613. [Google Scholar] [CrossRef]
- Duan, X.H.; Li, W.P.; Pei, C.H.; Zhou, X.Q. Molecular dynamics simulations of void defects in the energetic material HMX. J. Mol. Model. 2013, 19, 3893–3899. [Google Scholar] [CrossRef]
- Guillevic, M.; Pichot, V.; Cooper, J.; Coquerel, G.; Borne, L.; Spitzer, D. Optimization of an Antisolvent Method for RDX Recrystallization: Influence on Particle Size and Internal Defects. Cryst. Growth Des. 2019, 20, 130–138. [Google Scholar] [CrossRef]
- Dreger, Z.A.; Gupta, Y.M. Raman spectroscopy of high-pressure− high-temperature polymorph of hexahydro-1, 3, 5-trinitro-1, 3, 5-triazine (ε-RDX). J. Phys. Chem. A 2010, 114, 7038–7047. [Google Scholar] [CrossRef]
- Sabatini, J.J.; Oyler, K.D. Recent Advances in the Synthesis of High Explosive Materials. Crystals 2015, 6, 5. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Chen, D.; Yu, Y.; Li, H. Solubility of Octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine in 12 Pure Organic Solvents from 298.15 to 358.15 K. J. Chem. Eng. Data 2020, 65, 2098–2108. [Google Scholar] [CrossRef]
- Chen, F.; Zhou, T.; Wang, M. Spheroidal crystal morphology of RDX in mixed solvent systems predicted by molecular dynamics. J. Phys. Chem. Solids 2019, 136, 109196. [Google Scholar] [CrossRef]
- Zhang, C.; Ji, C.; Li, H.; Zhou, Y.; Xu, J.; Xu, R.; Li, J.; Luo, Y. Occupancy Model for Predicting the Crystal Morphologies Influenced by Solvents and Temperature, and Its Application to Nitroamine Explosives. Cryst. Growth Des. 2013, 13, 282–290. [Google Scholar] [CrossRef]
- Kim, C.-K.; Lee, B.-C.; Lee, Y.-W.; Kim, H.S. Solvent effect on particle morphology in recrystallization of HMX (cyclotetramethylenetetranitramine) using supercritical carbon dioxide as antisolvent. Korean J. Chem. Eng. 2009, 26, 1125–1129. [Google Scholar] [CrossRef]
- Sivabalan, R.; Gore, G.; Nair, U.; Saikia, A.; Venugopalan, S.; Gandhe, B. Study on ultrasound assisted precipitation of CL-20 and its effect on morphology and sensitivity. J. Hazard. Mater. 2007, 139, 199–203. [Google Scholar] [CrossRef]
- Sun, J.-K.; Sobolev, Y.I.; Zhang, W.; Zhuang, Q.; Grzybowski, B.A. Enhancing crystal growth using polyelectrolyte solutions and shear flow. Nature 2020, 579, 73–79. [Google Scholar] [CrossRef]
- Mao, X.; Song, X.; Lu, G.; Sun, Y.; Xu, Y.; Yu, J. Control of Crystal Morphology and Size of Calcium Sulfate Whiskers in Aqueous HCl Solutions by Additives: Experimental and Molecular Dynamics Simulation Studies. Ind. Eng. Chem. Res. 2015, 54, 4781–4787. [Google Scholar] [CrossRef]
- Taborga, P.; Brito, I.; Graber, T. Effect of additives on size and shape of lithium carbonate crystals. J. Cryst. Growth 2017, 460, 5–12. [Google Scholar] [CrossRef]
- Parambil, J.V.; Poornachary, S.K.; Tan, R.B.; Heng, J.Y. Influence of solvent polarity and supersaturation on template-induced nucleation of carbamazepine crystal polymorphs. J. Cryst. Growth 2017, 469, 84–90. [Google Scholar] [CrossRef]
- Cai, Z.; Liu, Y.; Song, Y.; Guan, G.; Jiang, Y. The effect of tailor-made additives on crystal growth of methyl paraben: Experiments and modelling. J. Cryst. Growth 2017, 461, 1–9. [Google Scholar] [CrossRef]
- Damiri, S.; Namvar, S.; Panahi, H. Micro-seeding and soft template effects on the control of polymorph and morphology of HMX micro particles in solvent-antisolvent process. Def. Technol. 2017, 13, 392–396. [Google Scholar] [CrossRef]
- Li, D.; Cai, J.; Guo, B.; Wei, N.; Liu, Q.; Liu, B. Controllable crystallization of urea crystal face (0 0 1) by molecular simulation. J. Cryst. Growth 2019, 524, 125139. [Google Scholar] [CrossRef]
- Lin, C.; Zeng, C.; Wen, Y.; Gong, F.; He, G.; Li, Y.; Yang, Z.; Ding, L.; Li, J.; Guo, S. Litchi-like Core–Shell HMX@HPW@PDA Microparticles for Polymer-Bonded Energetic Composites with Low Sensitivity and High Mechanical Properties. ACS Appl. Mater. Interfaces 2019, 12, 4002–4013. [Google Scholar] [CrossRef] [PubMed]
- Kosareva, E.K.; Zharkov, M.N.; Meerov, D.B.; Gainutdinov, R.V.; Fomenkov, I.V.; Zlotin, S.G.; Pivkina, A.N.; Kuchurov, I.V.; Muravyev, N.V. HMX surface modification with polymers via sc-CO2 antisolvent process: A way to safe and easy-to-handle energetic materials. Chem. Eng. J. 2021, 428, 131363. [Google Scholar] [CrossRef]
- Hartman, P.; Bennema, P. The attachment energy as a habit controlling factor. J. Cryst. Growth 1980, 49, 145–156. [Google Scholar] [CrossRef]
- Hartman, P. The attachment energy as a habit controlling factor: III. Application to corundum. J. Cryst. Growth 1980, 49, 166–170. [Google Scholar] [CrossRef]
- Duan, X.; Wei, C.; Liu, Y.; Pei, C. A molecular dynamics simulation of solvent effects on the crystal morphology of HMX. J. Hazard. Mater. 2010, 174, 175–180. [Google Scholar] [CrossRef]
- Liu, Y.; Niu, S.; Lai, W.; Yu, T.; Ma, Y.; Gao, H.; Zhao, F.; Ge, Z. Crystal morphology prediction of energetic materials grown from solution: Insights into the accurate calculation of attachment energies. CrystEngComm 2019, 21, 4910–4917. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, T.; Lai, W.; Ma, Y.; Cao, Y.; Liu, N.; Ge, Z.; Zhao, F. Deciphering Solvent Effect on Crystal Growth of Energetic Materials for Accurate Morphology Prediction. Cryst. Growth Des. 2020, 20, 521–524. [Google Scholar] [CrossRef]
- Yan, T.; Wang, J.-H.; Liu, Y.-C.; Zhao, J.; Yuan, J.-M.; Guo, J.-H. Growth and morphology of 1,3,5,7-tetranitro-1,3,5,7-tetraazacy-clooct ane (HMX) crystal. J. Cryst. Growth 2015, 430, 7–13. [Google Scholar] [CrossRef]
- Chen, G.; Xia, M.; Lei, W.; Wang, F.; Gong, X. Prediction of Crystal Morphology of Cyclotrimethylene Trinitramine in the Solvent Medium by Computer Simulation: A Case of Cyclohexanone Solvent. J. Phys. Chem. A 2014, 118, 11471–11478. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Xia, M.; Lei, W.; Wang, F.; Gong, X. Molecular dynamics investigation of the effect of solvent adsorption on crystal habits of hexogen. Can. J. Chem. 2014, 92, 849–854. [Google Scholar] [CrossRef]
- Choi, C.S.; Boutin, H.P. A study of the crystal structure of β-cyclotetramethylene tetranitramine by neutron diffraction. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1970, 26, 1235–1240. [Google Scholar] [CrossRef]
- Sun, H. COMPASS: An ab Initio Force-Field Optimized for Condensed-Phase ApplicationsOverview with Details on Alkane and Benzene Compounds. J. Phys. Chem. B 1998, 102, 7338–7364. [Google Scholar] [CrossRef]
- Li, J.; Jin, S.; Lan, G.; Xu, Z.; Wu, N.; Chen, S.; Li, L. The effect of solution conditions on the crystal morphology of β-HMX by molecular dynamics simulations. J. Cryst. Growth 2019, 507, 38–45. [Google Scholar] [CrossRef]
- Lei, W.; Dong, C.; Li, H.-Z.; Duan, X.-H.; Yu, Y.-W. Crystal Morphology of β—HMX under Eight Solvents System Using Molecular Dynamics Simulation and Experiment. Chinese J. Energetic Mater. 2020, 28, 317–329. [Google Scholar] [CrossRef]
- Wang, L. Study on the crystallization thermodynamics and crystal morphology control of HMX. Ph.D. Thesis, North University of China, Taiyuan, China, 2020. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abox/Å2 | a/Å | b/Å | c/Å | U × V | Thickness | |
|---|---|---|---|---|---|---|
| {0 1 1} | 1237.69 | 38.54 | 32.34 | 23.86 | 3 × 5 | 4 |
| {0 2 0} | 1209.71 | 38.17 | 32.34 | 21.12 | 5 × 5 | 4 |
| {1 1 −1} | 1388.17 | 38.17 | 36.57 | 22.27 | 5 × 3 | 4 |
| {1 0 0} | 1482.84 | 41.33 | 35.88 | 23.98 | 4 × 4 | 4 |
| {1 0 −2} | 1357.16 | 41.33 | 32.83 | 21.39 | 4 × 3 | 5 |
| hkl | dhkl/Å | Surface Area/Å2 | Eatt(Total)/kcal·mol−1 | Multiplicity | % Total Facet Area |
|---|---|---|---|---|---|
| {0 1 1} | 6.03 | 86.21 | −23.35 | 4 | 62.28 |
| {0 2 0} | 5.53 | 47.00 | −34.80 | 2 | 5.55 |
| {1 1 −1} | 5.52 | 94.04 | −36.58 | 4 | 29.83 |
| {1 0 0} | 5.40 | 96.14 | −48.64 | 2 | 1.26 |
| {1 0 −2} | 4.32 | 120.30 | −39.97 | 2 | 1.08 |
| hkl | Aacc/Å2 | Ahkl/Å2 | S |
|---|---|---|---|
| {0 1 1} | 111.90 | 81.54 | 1.37 |
| {0 2 0} | 65.64 | 47.23 | 1.39 |
| {1 1 −1} | 134.10 | 91.74 | 1.46 |
| {1 0 0} | 192.21 | 85.94 | 2.24 |
| {1 0 −2} | 144.01 | 107.58 | 1.34 |
| Ratio | E/kcal·mol−1 | {0 1 1} | {1 1 −1} | {1 0 0} | {0 2 0} | {1 0 −2} |
|---|---|---|---|---|---|---|
| 0.00 | Etot | −30,766.54 | −30,578.96 | −32,430.80 | −25,877.68 | −30,658.02 |
| Esur | −28,283.68 | −28,068.28 | −29,823.86 | −23,358.82 | −28,163.40 | |
| Esol | −2237.60 | −2206.11 | −2106.37 | −2238.73 | −2213.12 | |
| Eint | −245.26 | −304.57 | −500.57 | −280.13 | −281.50 | |
| Eatt-s | −7.04 | −9.15 | 9.52 | −24.86 | −17.64 | |
| Area percent (%) | 60.16 | 31.05 | 8.79 | 0.00 | 0.00 | |
| Aspect ratio | 1.80 | Volume/Å3 | 3314.45 | |||
| 0.05 | Etot | −31,352.37 | −31,214.67 | −33,032.86 | −26,434.76 | −31,265.86 |
| Esur | −28,484.00 | −28,323.34 | −30,043.34 | −23,557.29 | −28,370.54 | |
| Esol | −2603.56 | −2572.19 | −2460.07 | −2579.37 | −2591.87 | |
| Eint | −264.81 | −319.14 | −529.45 | −298.10 | −303.45 | |
| Eatt-s | −5.20 | −7.65 | 13.39 | −23.86 | −15.32 | |
| Area percent (%) | 67.21 | 32.79 | 0.00 | 0.00 | 0.00 | |
| Aspect ratio | 2.36 | Volume/Å3 | 1663.01 | |||
| 0.10 | Etot | −31,813.00 | −31,698.93 | −33,477.33 | −26,942.03 | −31,715.94 |
| Esur | −28,484.94 | −28,317.79 | −30,044.89 | −23,576.53 | −28,371.33 | |
| Esol | −3057.15 | −3041.24 | −2891.98 | −3072.08 | −3024.44 | |
| Eint | −270.91 | −339.90 | −540.46 | −293.42 | −320.17 | |
| Eatt-s | −4.62 | −5.50 | 14.86 | −24.12 | −13.56 | |
| Area percent (%) | 59.80 | 40.20 | 0.00 | 0.00 | 0.00 | |
| Aspect ratio | 2.08 | Volume/Å3 | 889.45 | |||
| 0.15 | Etot | −32,383.64 | −32,222.03 | −34,042.98 | −27,493.64 | −32,260.99 |
| Esur | −28,480.75 | −28,315.91 | −30,043.96 | −23,564.87 | −28,365.82 | |
| Esol | −3636.43 | −3549.40 | −3484.65 | −3632.15 | −3563.79 | |
| Eint | −266.46 | −356.72 | −514.37 | −296.62 | −331.38 | |
| Eatt-s | −5.04 | −3.76 | 11.37 | −23.94 | −12.38 | |
| Area percent (%) | 39.06 | 60.94 | 0.00 | 0.00 | 0.00 | |
| Aspect ratio | 2.26 | Volume/Å3 | 581.85 | |||
| 0.20 | Etot | −33,017.37 | −32,842.52 | −34,628.74 | −28,081.06 | −32,899.70 |
| Esur | −28,490.71 | −28,319.10 | −30,037.15 | −23,566.42 | −28,379.70 | |
| Esol | −4245.34 | −4178.96 | −4069.64 | −4204.66 | −4197.7 | |
| Eint | −281.32 | −344.46 | −521.95 | −309.99 | −322.22 | |
| Eatt-s | −3.64 | −5.03 | 12.38 | −23.20 | −13.34 | |
| Area percent (%) | 65.17 | 34.83 | 0.00 | 0.00 | 0.00 | |
| Aspect ratio | 2.27 | Volume/Å3 | 527.47 |
| Ratio (%) | E/kcal·mol−1 | {0 1 1} | {1 1 −1} | {1 0 0} | {0 2 0} | {1 0 −2} |
|---|---|---|---|---|---|---|
| 0.5% | Etot | −30,880.20 | −30,740.17 | −32,542.29 | −26,010.75 | −30,775.20 |
| Esur | −28,348.79 | −28,180.18 | −29,887.28 | −23,454.63 | −28,223.37 | |
| Esol | −2287.13 | −2248.49 | −2152.38 | −2270.38 | −2271.17 | |
| Eint | −244.28 | −311.50 | −502.63 | −285.74 | −280.66 | |
| Eatt-s | −7.14 | −8.44 | 9.80 | −24.55 | −17.73 | |
| Area percent (%) | 57.19 | 36.23 | 6.59 | 0.00 | 0.00 | |
| Aspect ratio | 1.74 | Volume/Å3 | 3111.26 | |||
| 1% | Etot | −30,897.80 | −30,754.38 | −32,571.84 | −26,036.31 | −30,806.18 |
| Esur | −28,351.52 | −28,188.67 | −29,896.64 | −23,446.25 | −28,225.52 | |
| Esol | −2295.01 | −2256.41 | −2177.98 | −2306.54 | −2292.15 | |
| Eint | −251.26 | −309.30 | −497.22 | −283.52 | −288.51 | |
| Eatt-s | −6.48 | −8.66 | 9.07 | −24.67 | −16.90 | |
| Area percent (%) | 61.42 | 30.29 | 8.30 | 0.00 | 0.00 | |
| Aspect ratio | 1.83 | Volume/Å3 | 2688.31 | |||
| 3% | Etot | −31,059.37 | −30,913.49 | −32,729.16 | −26,188.59 | −30,948.01 |
| Esur | −28,347.40 | −28,189.51 | −29,895.77 | −23,443.74 | −28,230.69 | |
| Esol | −2456.37 | −2414.52 | −2337.52 | −2455.73 | −2426.53 | |
| Eint | −255.60 | −309.46 | −495.87 | −289.11 | −290.79 | |
| Eatt-s | −6.07 | −8.65 | 8.89 | −24.36 | −16.66 | |
| Area percent (%) | 63.71 | 28.05 | 8.24 | 0.00 | 0.00 | |
| Aspect ratio | 1.91 | Volume/Å3 | 2396.22 | |||
| 5% | Etot | −31,229.13 | −31,061.91 | −32,888.67 | −26,355.00 | −31,108.00 |
| Esur | −28,346.02 | −28,184.10 | −29,913.24 | −23,455.19 | −28,235.15 | |
| Esol | −2630.58 | −2568.76 | −2499.61 | −2611.79 | −2585.25 | |
| Eint | −252.53 | −309.05 | −475.82 | −288.01 | −287.60 | |
| Eatt-s | −6.36 | −8.69 | 6.21 | −24.42 | −17.00 | |
| Area percent (%) | 58.01 | 20.32 | 21.67 | 0.00 | 0.00 | |
| Aspect ratio | 1.90 | Volume/Å3 | 2200.81 | |||
| 10% | Etot | −31,579.84 | −31,416.35 | −33,214.59 | −26,710.83 | −31,479.70 |
| Esur | −28,346.54 | −28,185.63 | −29,899.95 | −23,450.45 | −28,226.09 | |
| Esol | −2977.29 | −2919.23 | −2835.49 | −2975.52 | −2962.76 | |
| Eint | −256.01 | −311.49 | −479.16 | −284.86 | −290.85 | |
| Eatt-s | −6.03 | −8.44 | 6.66 | −24.59 | −16.65 | |
| Area percent (%) | 60.19 | 22.20 | 17.61 | 0.00 | 0.00 | |
| Aspect ratio | 1.89 | Volume/Å3 | 2049.35 |
| hkl | {0 1 1} | {1 1 −1} | {1 0 0} | {0 2 0} | {1 0 −2} | |
|---|---|---|---|---|---|---|
| PAA | Etot | −31,230.07 | −31,065.25 | −32,896.09 | −26,323.79 | −31,104.88 |
| Esur | −28,352.24 | −28,177.58 | −29,896.12 | −23,452.25 | −28,232.23 | |
| Esol | −2626.28 | −2583.04 | −2478.41 | −2586.87 | −2591.39 | |
| Eint | −251.55 | −304.64 | −521.57 | −284.67 | −281.26 | |
| Eatt-s | −6.45 | −9.15 | 12.33 | −24.61 | −17.66 | |
| Area percent (%) | 65.52 | 32.79 | 1.69 | 0.00 | 0.00 | |
| Aspect ratio | 1.96 | Volume/Å3 | 3015.05 | |||
| PEI | Etot | −30,544.08 | −30,378.86 | −32,209.80 | −25,669.36 | −30,455.21 |
| Esur | −28,346.89 | −28,185.26 | −29,898.30 | −23,447.44 | −28,203.71 | |
| Esol | −1934.56 | −1899.78 | −1813.94 | −1938.37 | −1903.81 | |
| Eint | −262.64 | −293.82 | −497.56 | −283.56 | −347.69 | |
| Eatt-s | −5.40 | −10.26 | 9.12 | −24.67 | −10.66 | |
| Area percent (%) | 71.43 | 16.20 | 10.80 | 0.00 | 1.58 | |
| Aspect ratio | 2.24 | Volume/Å3 | 2317.52 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhai, L.; Zhao, X.; Rui, J.; Qiu, S.; Li, Y. Molecular Dynamics Simulation of β-HMX Crystal Morphology Induced by Polymer Additives. Crystals 2022, 12, 164. https://doi.org/10.3390/cryst12020164
Zhai L, Zhao X, Rui J, Qiu S, Li Y. Molecular Dynamics Simulation of β-HMX Crystal Morphology Induced by Polymer Additives. Crystals. 2022; 12(2):164. https://doi.org/10.3390/cryst12020164
Chicago/Turabian StyleZhai, Lizhe, Xue Zhao, Jiuhou Rui, Siqi Qiu, and Yongshen Li. 2022. "Molecular Dynamics Simulation of β-HMX Crystal Morphology Induced by Polymer Additives" Crystals 12, no. 2: 164. https://doi.org/10.3390/cryst12020164