
Temperature-Resolved Anisotropic Displacement Parameters from Theory and Experiment: A Case Study


 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
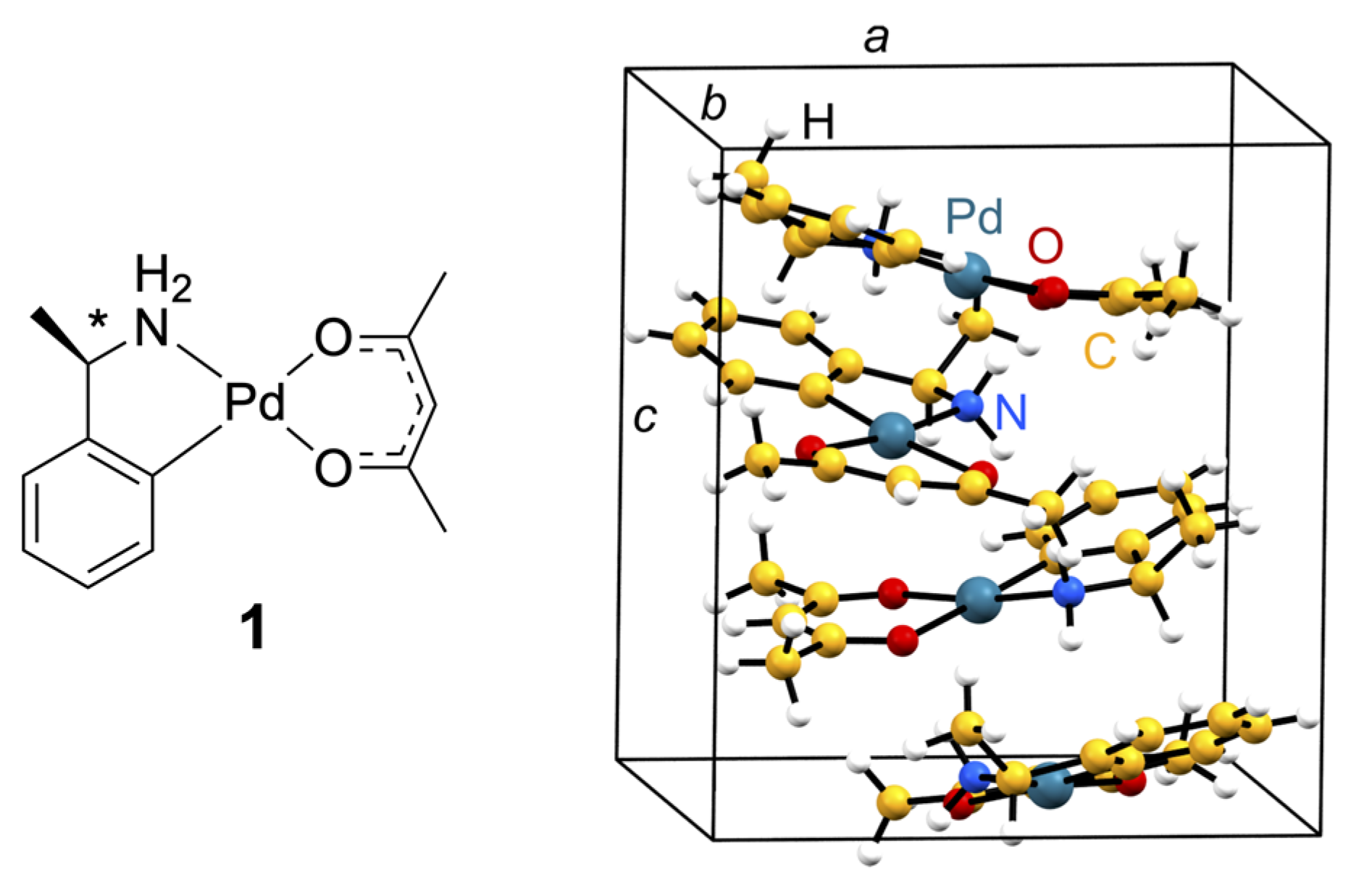
2. Materials and Methods
2.1. Diffraction Experiments
2.2. Theoretical Calculations
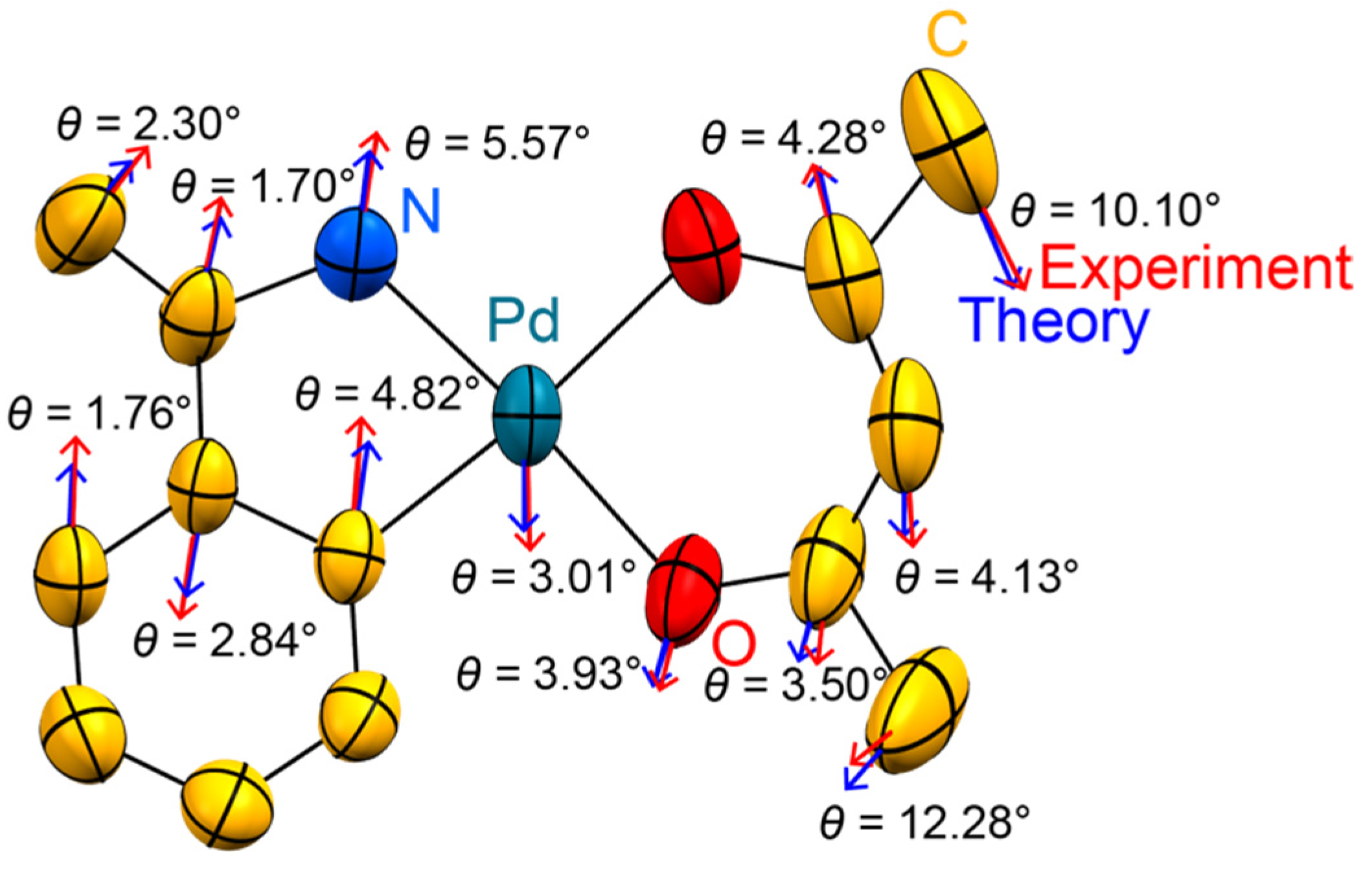
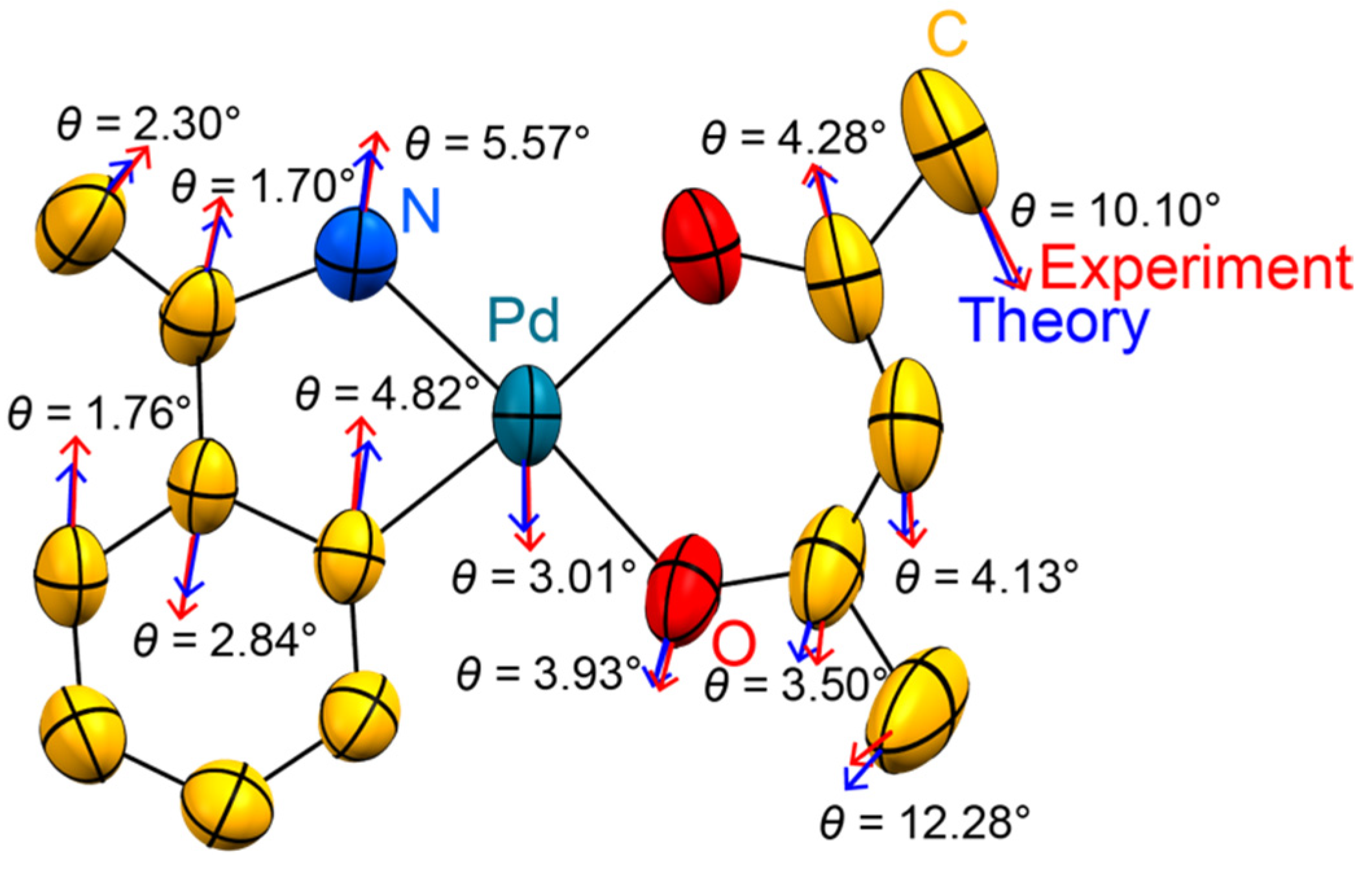
3. Results
4. Discussion
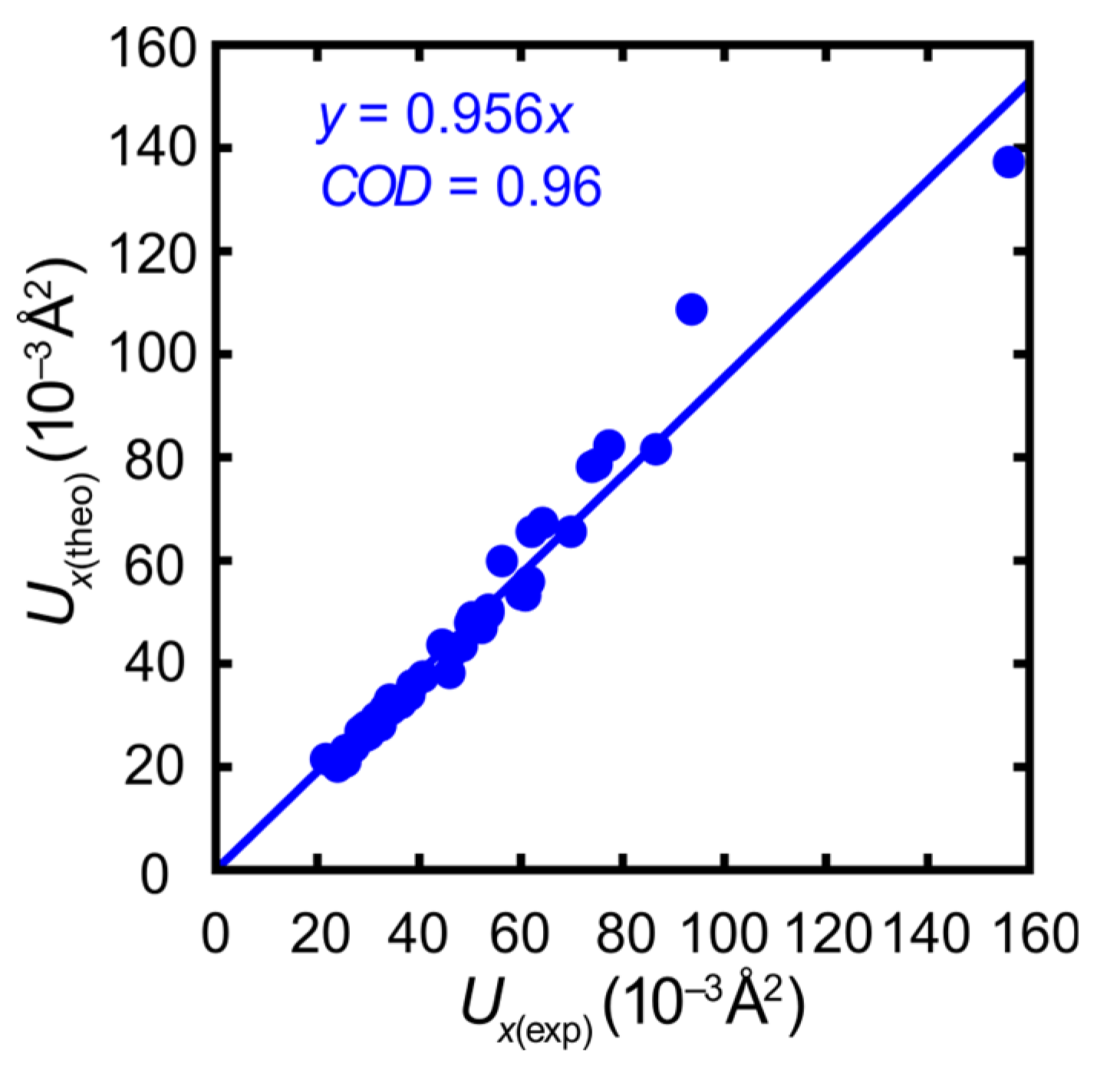
- Neither the individual sample nor the details of the instrumentation are reflected in drastically different ADPs. The directionality of the most prolate ADPs matches that from the temperature-dependent series (Figure S2 in the Supplementary Material). The ADP amplitudes derived from the control experiment are reasonably close to those expected by extrapolating the data collections on the first crystal to 100 K.
- The high resolution of the control is comforting for a benchmark experiment but not mandatory. The truncation of this highly redundant data set to the IUCr standard resolution of 0.6 Å−1 only leads to an insignificant change of less than 1%.
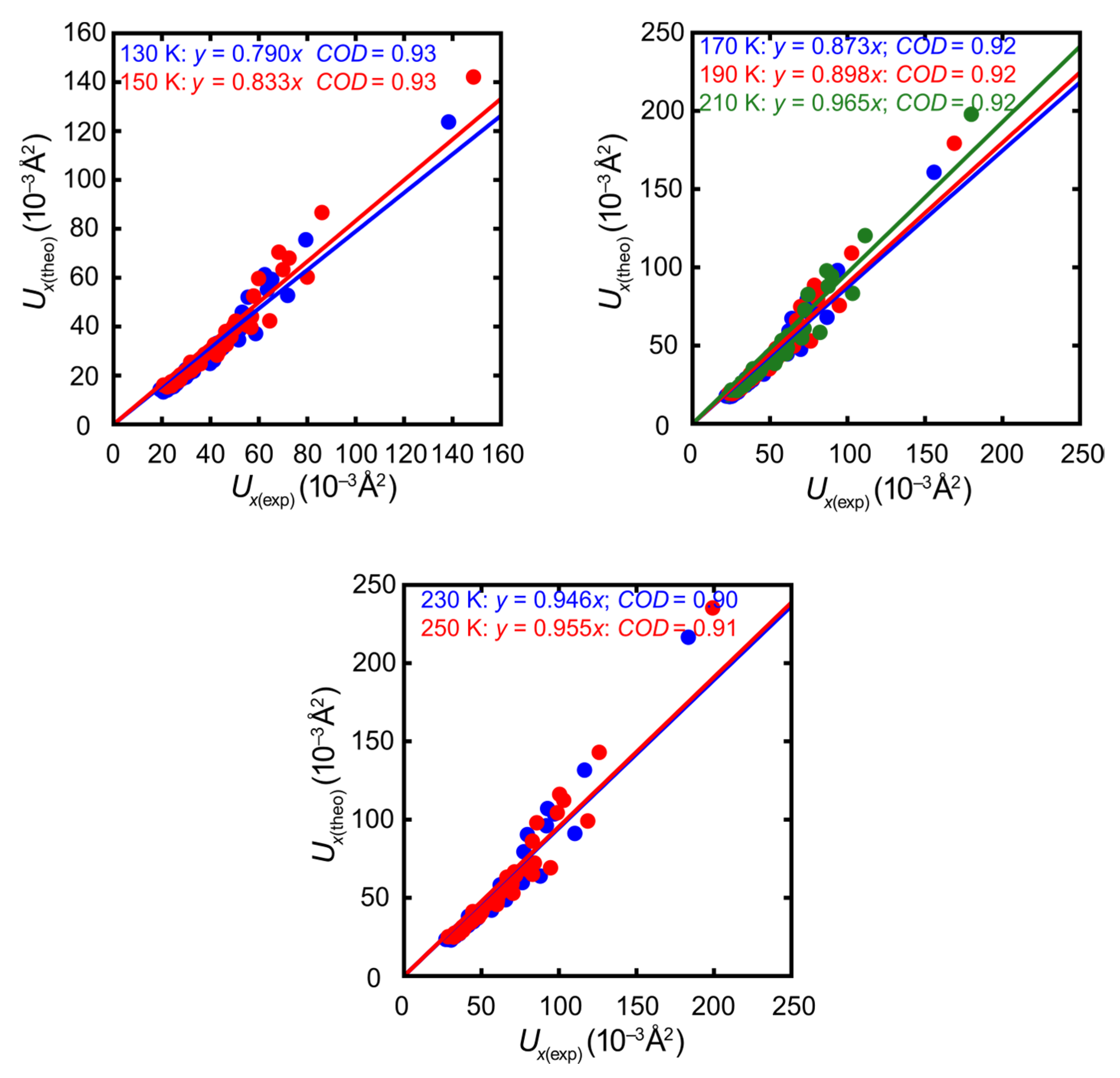
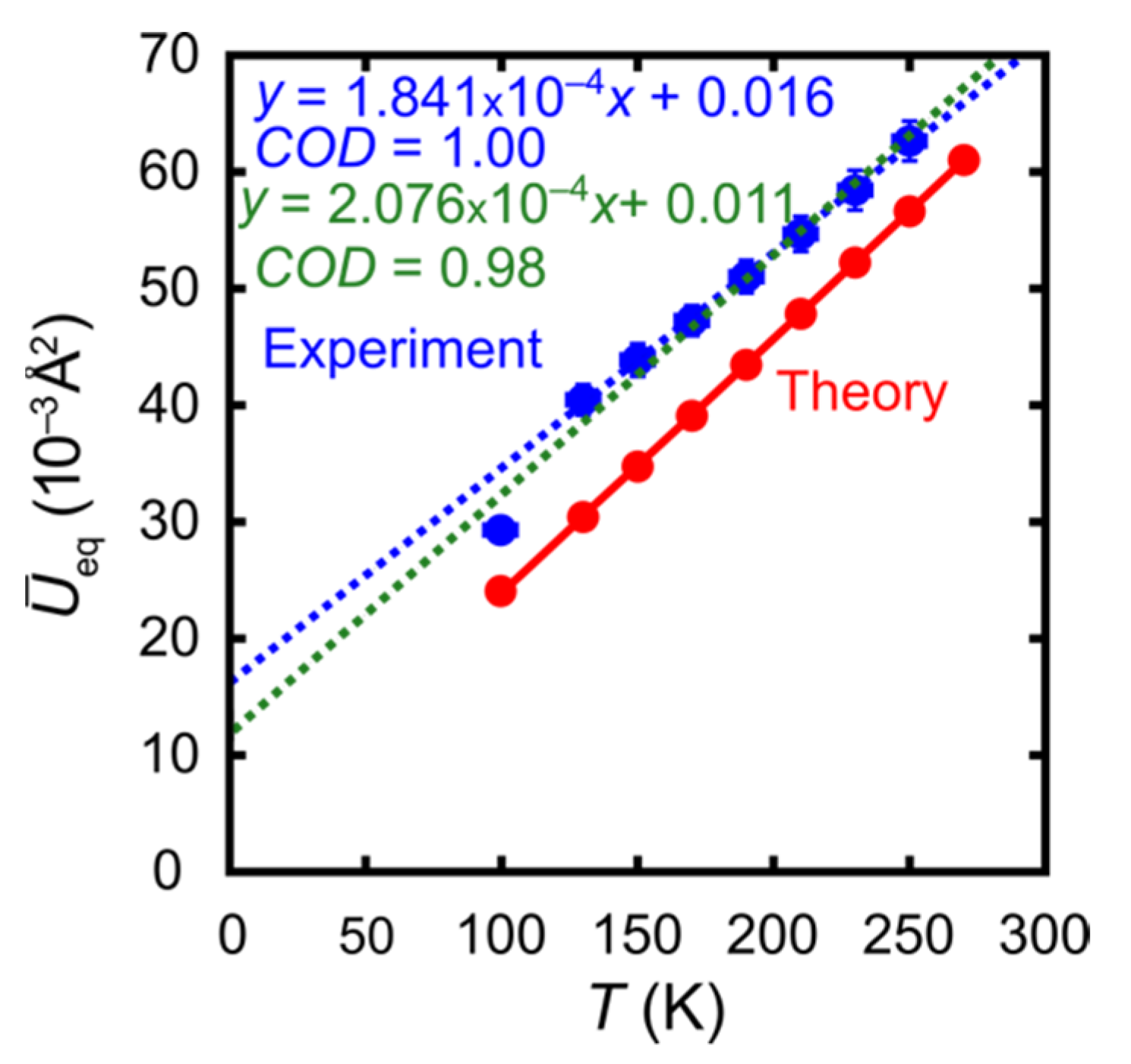
- The synopsis of both experiments—temperature-dependent diffraction on the first crystal and high-resolution diffraction on the second crystal—provides a more realistic picture of experimental errors than the numerical standard uncertainties of the refined ADP components. The discrepancy between the alternative correlations represented by the blue and green dashed lines in Figure 5 is modest but clearly visible. In other words, even the ADPs from carefully conducted experiments differ more than their numerical standard uncertainties suggest, and theory matches almost as well as an alternative experiment.
5. Conclusions and Future Work
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- George, J.; Deringer, V.L.; Wang, A.; Müller, P.; Englert, U.; Dronskowski, R. Lattice thermal expansion and anisotropic displacements in α-sulfur from diffraction experiments and first-principles theory. J. Chem. Phys. 2016, 145, 234512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, J.; Wang, R.; Englert, U.; Dronskowski, R. Lattice thermal expansion and anisotropic displacements in urea, bromomalonic aldehyde, pentachloropyridine, and naphthalene. J. Chem. Phys. 2017, 147, 074112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, J.; Wang, A.; Deringer, V.L.; Wang, R.; Dronskowski, R.; Englert, U. Anisotropic displacement parameters from dispersion-corrected DFT methods and their experimental validation by temperature-dependent X-ray diffraction. CrystEngComm 2015, 17, 7414–7422. [Google Scholar] [CrossRef] [Green Version]
- Schwarzenbach, D. The success story of crystallography. Z. Kristallogr. Cryst. Mater. 2012, 227, 52–62. [Google Scholar] [CrossRef]
- Glusker, J.P.; Lewis, M.; Rossi, M. Crystal Structure Analysis for Chemists and Biologists, 1st ed.; VCH Publishers: New York, NY, USA, 1994. [Google Scholar]
- Mroz, D.; Wang, R.; Englert, U.; Dronskowski, R. Can we trust the experiment? Anisotropic displacement parameters in 1-(halogenmethyl)-3-nitrobenzene (halogen = Cl, Br). Acta Crystallogr. Sect. C 2020, 76, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Calmuschi, B.; Englert, U. (S)-(Acetylacetonato-κ2O,O)[2-(1-aminoethyl)phenyl-κ2C1,N]palladium(II). Acta Crystallogr. Sect. E 2005, 61, m164–m165. [Google Scholar] [CrossRef]
- P24 Chemical Crystallography. Available online: https://photon-science.desy.de/facilities/petra_iii/beamlines/p24_chemical_crystallography/index_eng.html (accessed on 16 January 2022).
- Bruker. SADABS; Bruker: Madison, WI, USA, 2014. [Google Scholar]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Togo, A.; Oba, F.; Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Phys. Rev. B 2008, 78, 134106. [Google Scholar] [CrossRef] [Green Version]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Deringer, V.L.; Stoffel, R.P.; Togo, A.; Eck, B.; Meven, M.; Dronskowski, R. Ab initio ORTEP drawings: A case study of N-based molecular crystals with different chemical nature. CrystEngComm 2014, 16, 10907–10915. [Google Scholar] [CrossRef]
- Parlinski, K.; Li, Z.Q.; Kawazoe, Y. First-Principles Determination of the Soft Mode in Cubic ZrO2. Phys. Rev. Lett. 1997, 78, 4063–4066. [Google Scholar] [CrossRef]
- Grosse-Kunstleve, R.W.; Adams, P.D. On the handling of atomic anisotropic displacement parameters. J. Appl. Crystallogr. 2002, 35, 477–480. [Google Scholar] [CrossRef] [Green Version]
- George, J. Molecular Toolbox and Additional Information Regarding ADP Computation; Ellipsoids; Available online: http://www.ellipsoids.de (accessed on 16 January 2022).
- MATLAB. R 2016b; The MathWorks Inc.: Natick, MA, USA, 2016. [Google Scholar]
- George, J.; Deringer, V.L.; Dronskowski, R. Dimensionality of Intermolecular Interactions in Layered Crystals by Electronic-Structure Theory and Geometric Analysis. Inorg. Chem. 2015, 54, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, R.P.; Wessel, C.; Lumey, M.W.; Dronskowski, R. Ab initio thermochemistry of solid-state materials. Angew. Chem. Int. Ed. 2010, 49, 5242–5266. [Google Scholar] [CrossRef] [PubMed]
- Erba, A.; Maul, J.; Itou, M.; Dovesi, R.; Sakurai, Y. Anharmonic Thermal Oscillations of the Electron Momentum Distribution in Lithium Fluoride. Phys. Rev. Lett. 2015, 115, 117402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinet, P.; Smith, J.R.; Ferrante, J.; Rose, J.H. Temperature effects on the universal equation of state of solids. Phys. Rev. B 1987, 35, 1945–1953. [Google Scholar] [CrossRef] [Green Version]
- Mroz, D.; George, J.; Kremer, M.; Wang, R.; Englert, U.; Dronskowski, R. A new tool for validating theoretically derived anisotropic displacement parameters with experiment: Directionality of prolate displacement ellipsoids. CrystEngComm 2019, 21, 6396–6404. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Can X-ray data distinguish bonding effects from vibrational smearing? Acta Crystallogr. Sect. A 1976, 32, 239–244. [Google Scholar] [CrossRef]
- Wang, R.; Hartnick, D.; Englert, U. Short is strong: Experimental electron density in a very short N···I halogen bond. Z. Krist.-Cryst. Mater. 2018, 233, 733–744. [Google Scholar] [CrossRef]
- Wang, R.; George, J.; Potts, S.K.; Kremer, M.; Dronskowski, R.; Englert, U. The many flavours of halogen bonds–message from experimental electron density and Raman spectroscopy. Acta Crystallogr. Sect. C 2019, 75, 1190–1201. [Google Scholar] [CrossRef] [Green Version]
- Madsen, A.Ø.; Civalleri, B.; Ferrabone, M.; Pascale, F.; Erba, A. Anisotropic displacement parameters for molecular crystals from periodic Hartree–Fock and density functional theory calculations. Acta Crystallogr. Sect. A 2013, 69, 309–321. [Google Scholar] [CrossRef]
- Deringer, V.L.; Wang, A.; George, J.; Dronskowski, R.; Englert, U. Anisotropic thermal motion in transition-metal carbonyls from experiments and ab initio theory. Dalton Trans. 2016, 45, 13680–13685. [Google Scholar] [CrossRef] [Green Version]
- Willis, B.T.M.; Pryor, A.W. Thermal Vibrations in Crystallography, 1st ed.; Cambridge University Press: Cambridge, UK, 1975. [Google Scholar]
- Bürgi, H.B.; Capelli, S.C.; Birkedal, H. Anharmonicity in anisotropic displacement parameters. Acta Crystallogr. Sect. A 2000, 56, 425–435. [Google Scholar] [CrossRef]
- Lindemann, F.A. The Calculcation of Molecular Vibration Frequencies. Phys. Z. 1910, 11, 609–615. [Google Scholar]
- Löwen, H. Melting, freezing and colloidal suspensions. Phys. Rep. 1994, 237, 249–324. [Google Scholar] [CrossRef]
- Bürgi, H.B.; Förtsch, M. Dynamic processes and disorder in crystal structures as seen by temperature-dependent diffraction experiments. J. Mol. Struct. 1999, 485–486, 457–463. [Google Scholar] [CrossRef]
- Capelli, S.C.; Albinati, A.; Mason, S.A.; Willis, B.T.M. Molecular motion in crystalline naphthalene: Analysis of multi-temperature X-ray and neutron diffraction data. J. Phys. Chem. A 2006, 110, 11695–11703. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Merkens, C.; Si, R.; Englert, U. Crosslinking of the Pd(acacCN)2 building unit with Ag(I) salts: Dynamic 1D polymers and an extended 3D network. CrystEngComm 2015, 17, 4383–4393. [Google Scholar] [CrossRef] [Green Version]
- Strothmann, R.; van Terwingen, S.; Kalf, I.; Englert, U. Systematic screening for k type phase transitions–general approach and positive example for a binuclear Cu(II) paddlewheel structure. CrystEngComm 2021, 23, 841–849. [Google Scholar] [CrossRef]
- Albino, A.; Benci, S.; Atzori, M.; Chelazzi, L.; Ciattini, S.; Taschin, A.; Bartolini, P.; Lunghi, A.; Righini, R.; Torre, R.; et al. Temperature Dependence of Spin-Phonon Coupling in [VO(acac)2]: A Computational and Spectroscopic Study. J. Phys. Chem. C 2021, 125, 22100–22110. [Google Scholar] [CrossRef]
- Moseley, D.H.; Stavretis, S.E.; Thirunavukkuarasu, K.; Ozerov, M.; Cheng, Y.; Daemen, L.L.; Ludwig, J.; Lu, Z.; Smirnov, D.; Brown, C.M.; et al. Spin–phonon couplings in transition metal complexes with slow magnetic relaxation. Nat. Commun. 2018, 9, 2572. [Google Scholar] [CrossRef]
- Madsen, A.Ø. SHADE web server for estimation of hydrogen anisotropic displacement parameters. J. Appl. Crystallogr. 2006, 39, 757–758. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mroz, D.; Wang, R.; Paulmann, C.; Englert, U.; Dronskowski, R. Temperature-Resolved Anisotropic Displacement Parameters from Theory and Experiment: A Case Study. Crystals 2022, 12, 283. https://doi.org/10.3390/cryst12020283
Mroz D, Wang R, Paulmann C, Englert U, Dronskowski R. Temperature-Resolved Anisotropic Displacement Parameters from Theory and Experiment: A Case Study. Crystals. 2022; 12(2):283. https://doi.org/10.3390/cryst12020283
Chicago/Turabian StyleMroz, Damian, Ruimin Wang, Carsten Paulmann, Ulli Englert, and Richard Dronskowski. 2022. "Temperature-Resolved Anisotropic Displacement Parameters from Theory and Experiment: A Case Study" Crystals 12, no. 2: 283. https://doi.org/10.3390/cryst12020283
APA StyleMroz, D., Wang, R., Paulmann, C., Englert, U., & Dronskowski, R. (2022). Temperature-Resolved Anisotropic Displacement Parameters from Theory and Experiment: A Case Study. Crystals, 12(2), 283. https://doi.org/10.3390/cryst12020283