
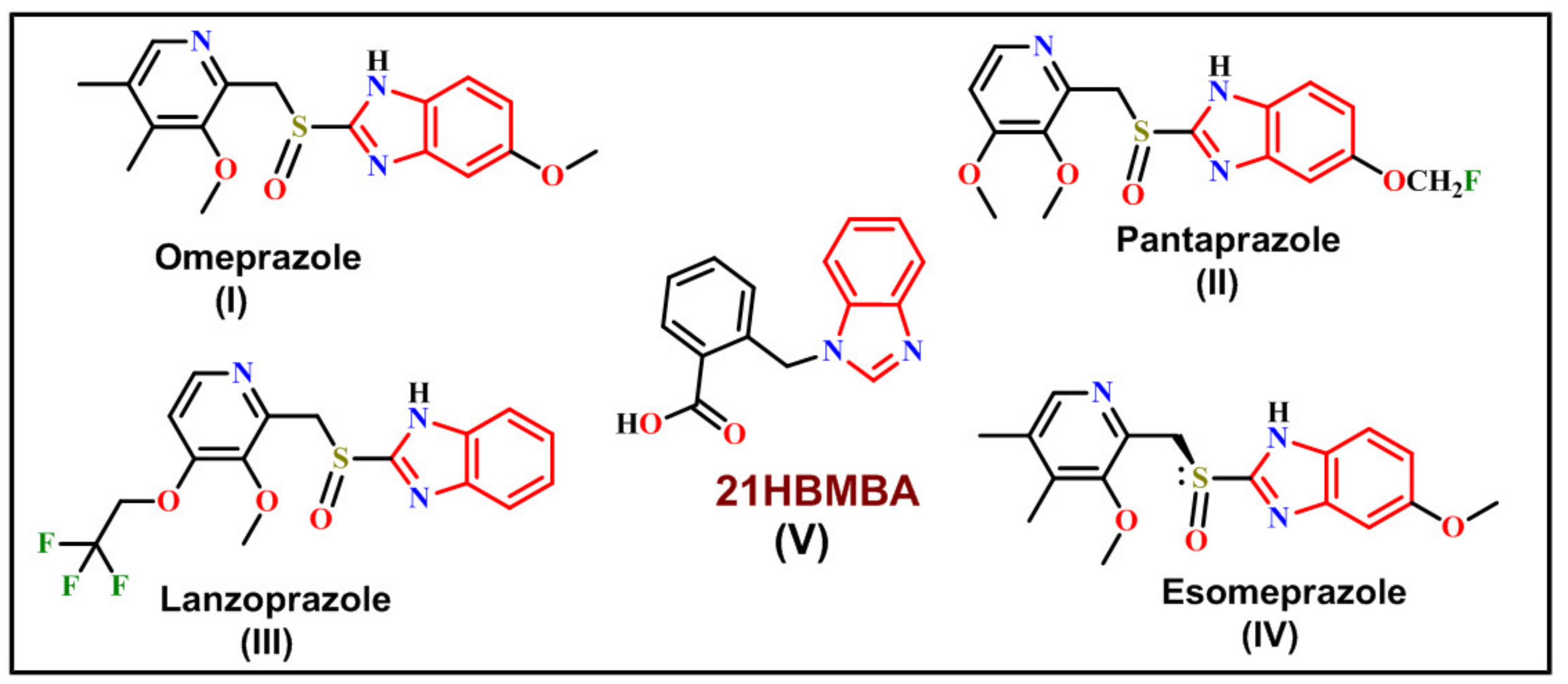
Vibrational Spectroscopy, Quantum Computational and Molecular Docking Studies on 2-[(1H-Benzimidazol-1-yl)-methyl]benzoic Acid

 , , ,
, , ,  ,
,
Abstract
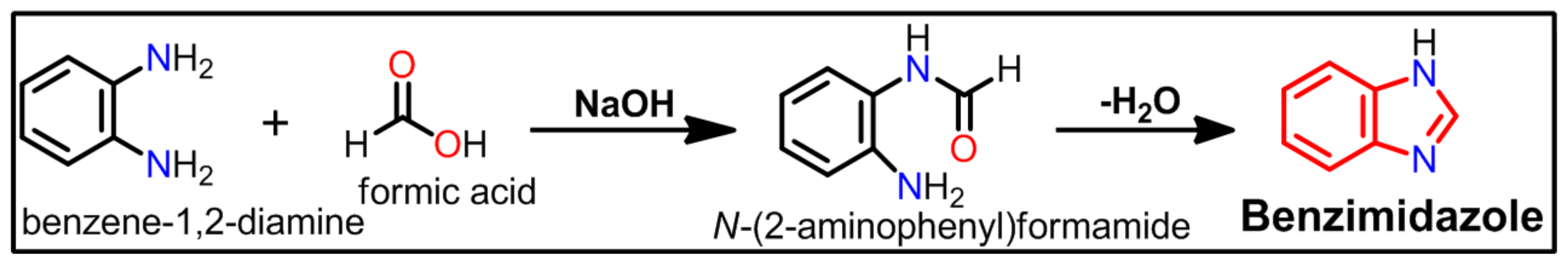
:1. Introduction
2. Material and Methods
2.1. Methods and Instrumentation
2.2. Computational Details
3. Results and Discussion
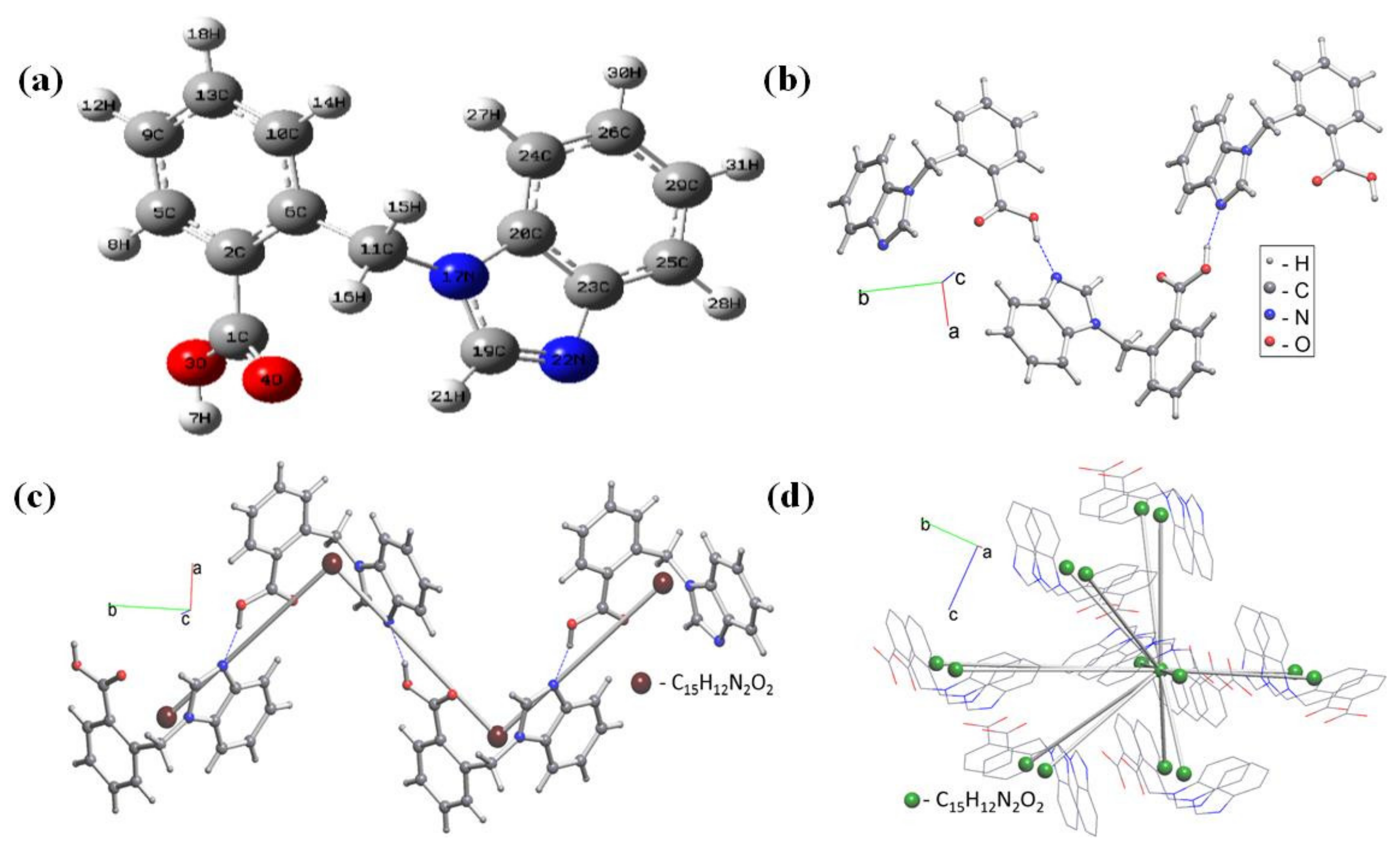
3.1. Optimized Molecular Geometry
3.2. Vibrational Spectroscopic Analysis
3.2.1. C–H Stretching Modes
3.2.2. C–C Vibrations
3.2.3. C–N Vibrations
3.2.4. C=O and O–H Vibrations
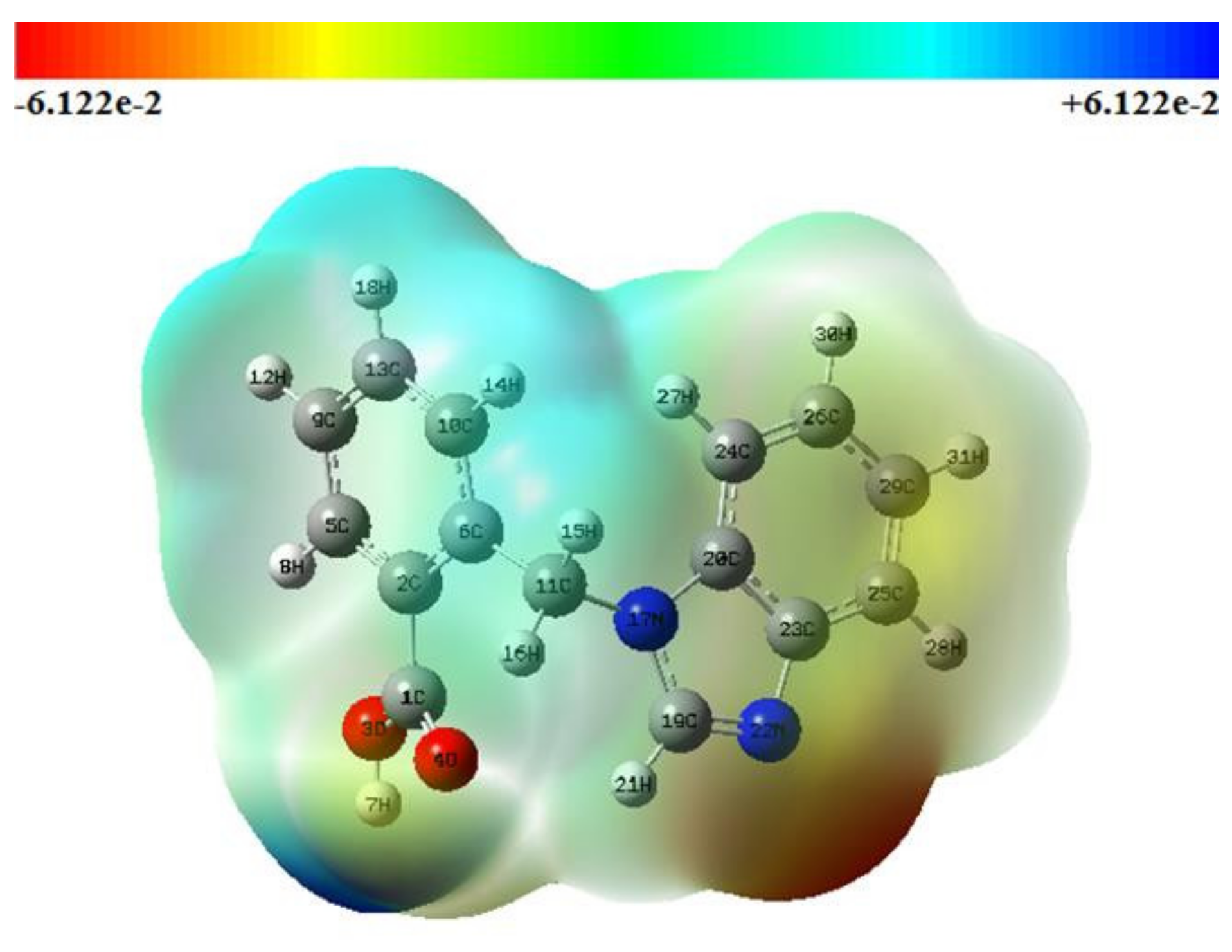
3.3. Molecular Electrostatic Potential (MEP)
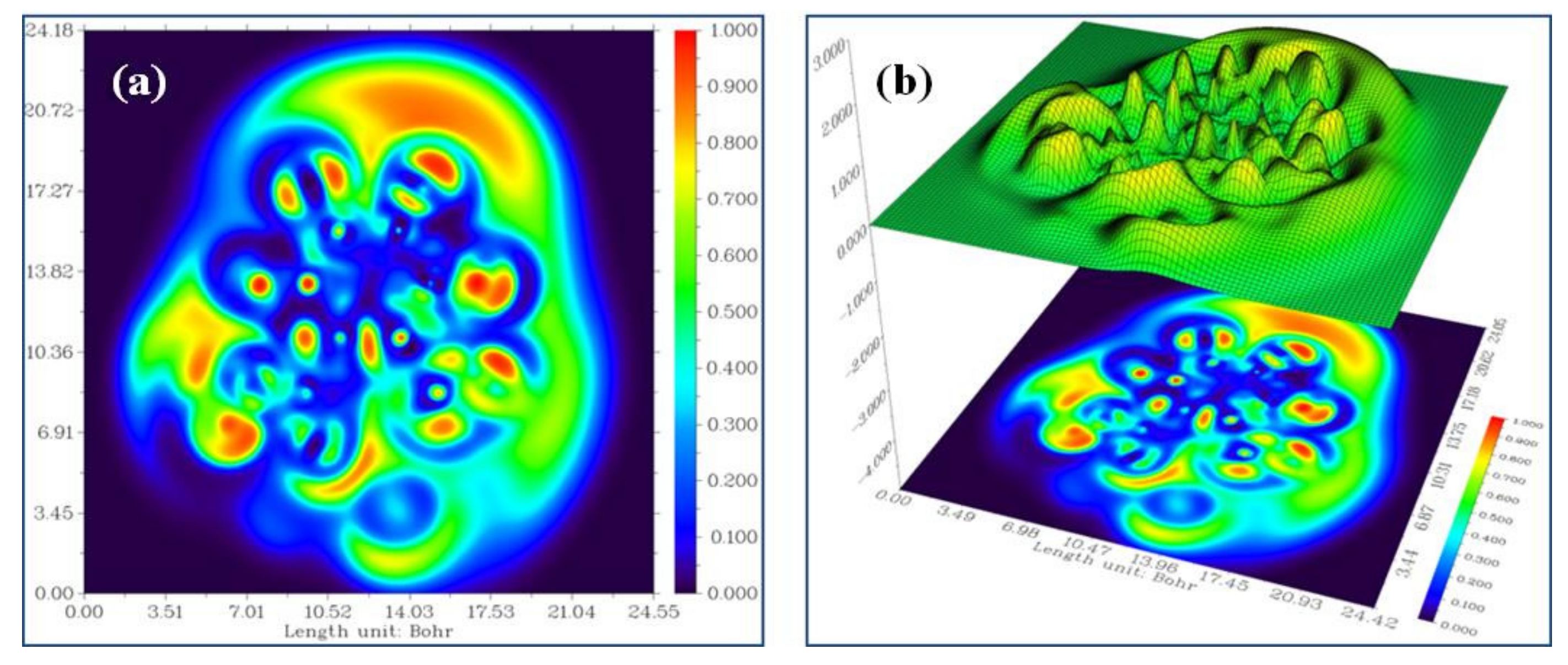
3.4. Electron Localization Function (ELF)
3.5. Non-Linear Optics
3.6. Donor Acceptor Interaction
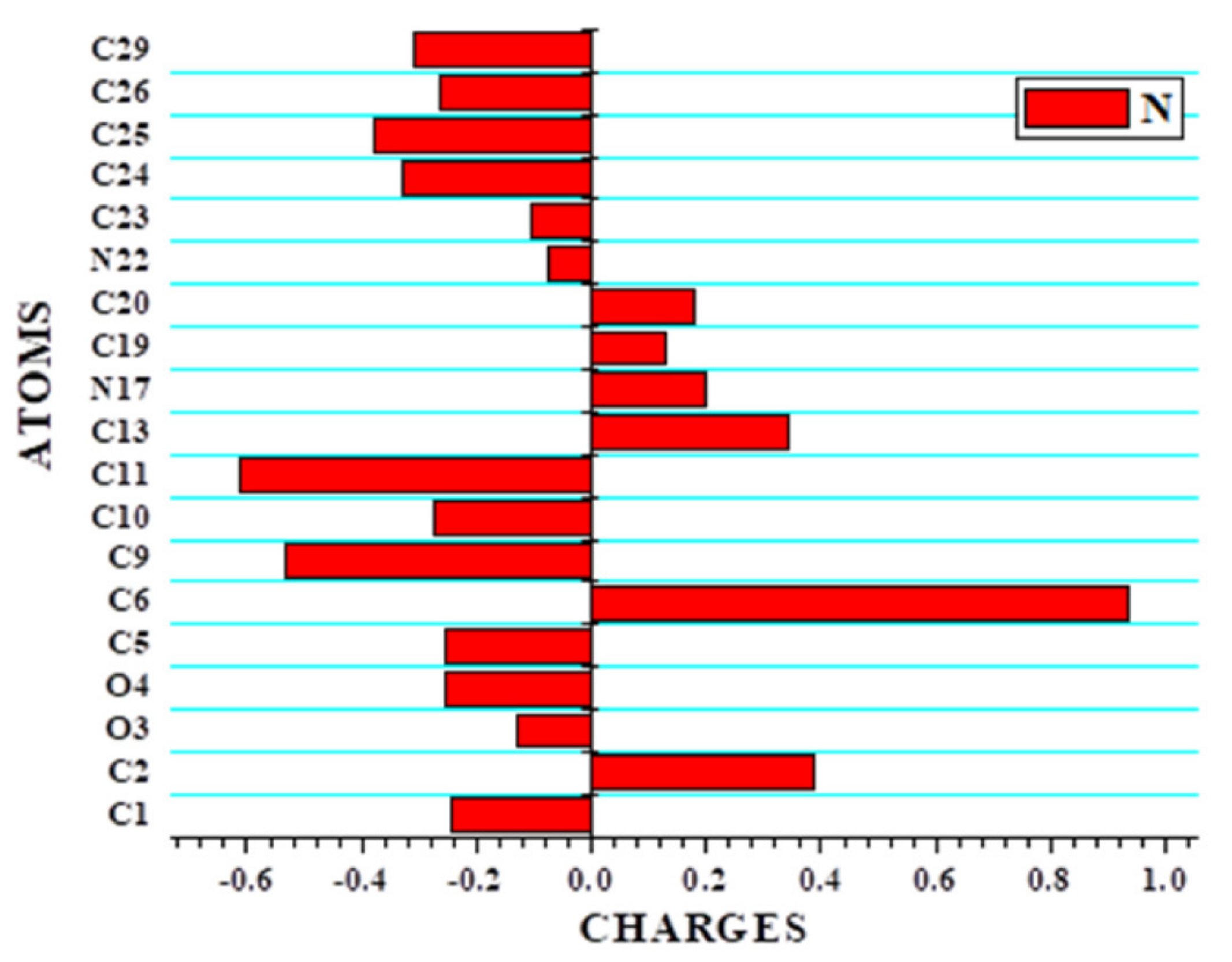
3.7. Population Analysis (Local Reactivity Descriptor: Fukui Function)
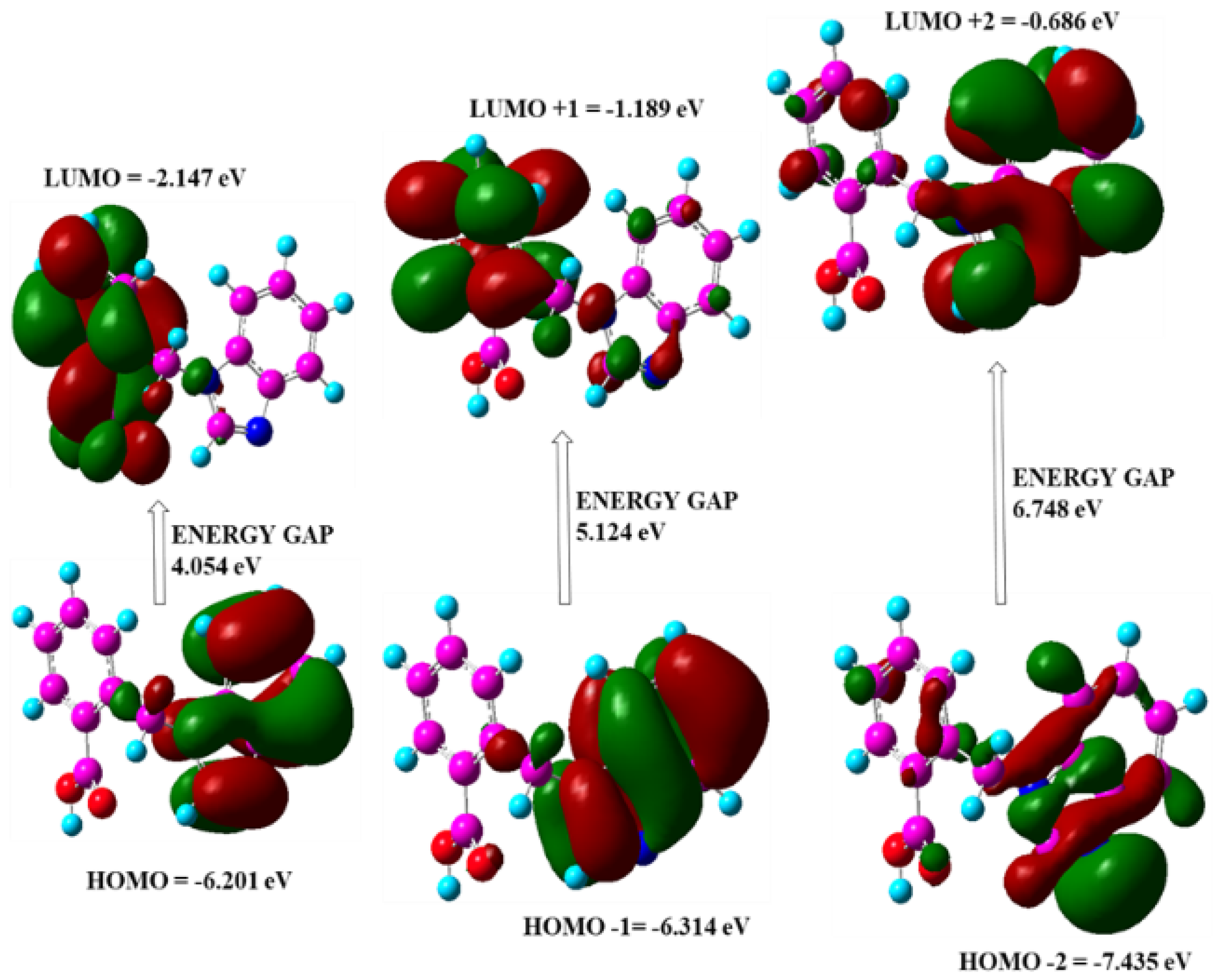
3.8. Frontier Molecular Orbital Analysis
3.9. 1HNMR Spectral Analysis
3.10. EDD and HDD Profiles for Excited States of 21HBMBA
3.11. Druglikeness
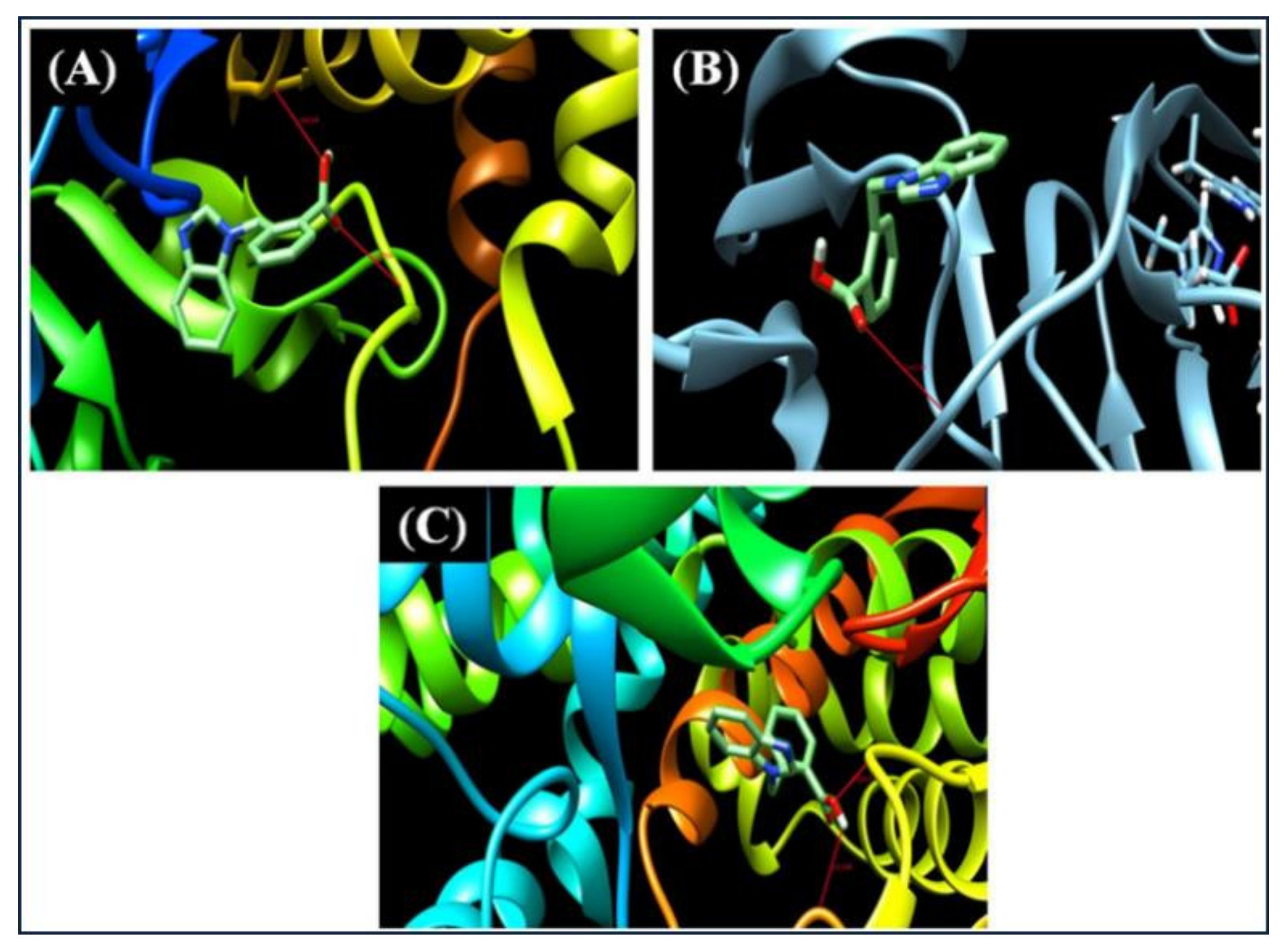
3.12. Molecular Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sivakumar, R.; Pradeepchandran, R.; Jayaveera, K.N.; Kumarnallasivan, P.; Vijaianand, P.R.; Venkatnarayanan, R. Benzimidazole: An attractive pharmacophore in medicinal chemistry. Int. J. Pharm. Res. 2011, 3, 19–31. [Google Scholar]
- Hobrecker, F. Reduction-products of nitracetamide compounds. Deut. Chem. Ges. Ber. 1872, 5, 920–924. [Google Scholar] [CrossRef] [Green Version]
- Ramanatham, V.; Sanjay, D.V.; Kumar, B.V.S.; Umesh, N.B.; Shekar, B.B.; Mashelkar, U.C. Synthesis, anti-bacterial, anti-asthmatic and anti-diabetic activities of novel N-substituted 2-(4-styrylphenyl)-1H-benzimidazole and N- substituted-3[4-(1H-benzimidazole-2-yl)-phenyl]-acrylic acid tert-butyl ester. ARKIVOC 2008, 10, 37–49. [Google Scholar]
- Valdez, J.; Cedillo, R.; Hernandez-Campos, A.; Yepez, L.; Hernandez-Luis, F.; Navarrete-Vazquez, G.; Tapia, A.; Cortes, R.; Hernandezc, M.; Castilloa, R. Synthesis and antiparasitic activity of 1H-benzimidazole derivatives. Bioorg. Med. Chem. Lett. 2002, 12, 2221–2224. [Google Scholar] [CrossRef]
- Fonseca, T.; Gigante, B.; Gilchrist, T.L. A short synthesis of phenanthro[2,3-d]imidazoles from dehydroabietic acid, Application of the methodology as a convenient route to benzimidazoles. Tetrahedron 2001, 57, 1793–1799. [Google Scholar] [CrossRef]
- Pabba, C.; Wang, H.J.; Mulligan, S.R.; Chen, Z.J.; Stark, T.M.; Gregg, B.T. Microwave- assisted synthesis of 1-aryl-1H-indazolesvia one-pot two-step Cu-catalyzed intramolecular N-arylation of arylhydrazones. Tetrahedron Lett. 2005, 46, 7553–7557. [Google Scholar] [CrossRef]
- Denny, W.A.; Rewcastle, G.W.; Baguley, B.C. Potential antitumor agents. 59. structure-activity relationships for 2-phenylbenzimidazole-4-carboxamidesa, new class of “minimal” DNA-Intercalating agents which may not actvia topoisomerase II. J. Med. Chem. 1990, 33, 814–819. [Google Scholar] [CrossRef]
- Gomez, H.T.; Nunez, E.H.; Rivera, I.L.; Alvarez, J.G.; Rivera, R.C.; Puc, R.M.; Ramos, R.A.; Gutierrez, M.C.R.; Bacab, M.J.C.; Vazquez, G.N. Design, synthesis andin vitro antiprotozoal activity of benzimidazolepentamidine hybrids, Bioorg. Med. Chem. Lett. 2008, 18, 3147–3151. [Google Scholar] [CrossRef]
- Porcari, A.R.; Devivar, R.V.; Kucera, L.S.; Drach, J.C.; Townsend, L.B. Design, synthesis, and antiviral evaluations of 1-(substituted benzyl)-2-substituted-5,6-dichlorobenzimidazoles as nonnucleoside analogues of 2,5,6-trichloro-1-(β-Dribofuranosyl)benzimidazole. J. Med. Chem. 1998, 41, 1252–1262. [Google Scholar] [CrossRef]
- Tamm, I.; Sehgal, P.B. Halobenzimidazoleribosides and RNA synthesis of cells and viruses. Adv. Virus Res. 1978, 22, 187–258. [Google Scholar]
- Roth, M.; Morningstar, M.L.; Boyer, P.L.; Hughes, S.H.; Buckheit, R.W., Jr.; Michejda, C.J. Synthesis and biological activity of novel nonnucleoside inhibitors of HIV-1 reverse transcriptase 2-aryl-substituted benzimidazoles. J. Med. Chem. 1997, 40, 4199–4207. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.S.; Lopes, P.F. Injectable Formulation of a Macrocyclic Lactone and Levamisole. US Patent No. 20130090296, 23 June 2011. [Google Scholar]
- Stefanska, J.Z.; Gralewska, R.; Starosciak, B.J.; Kazimierczuk, Z. Antimicrobial activity of substituted azoles and their nucleosides. Pharmazie 1999, 54, 879–884. [Google Scholar]
- Boiani, M.; Gonzalez, M. Imidazole and benzimidazole derivatives as chemotherapeutic agents. Mini Rev. Med. Chem. 2005, 5, 409–424. [Google Scholar] [CrossRef]
- Warren, M.J. Finding the final pieces of the vitamin B12 biosynthetic jigsaw. Proc. Natl. Acad. Sci. USA 2006, 103, 4799–4800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bino, A.; Baldisserotto, A.; Scalambra, E.; Dissette, V.; Vedaldi, D.E.; Salvador, A.; Durini, E.; Manfredini, S.; Vertuani, S. Design, synthesis and biological evaluation of novel hydroxy-phenyl-1H-benzimidazoles as radical scavengers and UV-protective agents. J. Enzyme Inhib. Med. Chem. 2017, 32, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Dikshit, D.V.; Deval, S.D.; Deodhar, K.D. Synthesis of benzimidazole substituted pyridone azo disperse dyes. Dyes Pigments 1985, 6, 39–46. [Google Scholar] [CrossRef]
- Rangappa, S.K.; Hiremathad, A.; Budagumpi, S.; Nagaraja, B.M. Comprehensive Review in Current Developments of Benzimidazole—Based Medicinal Chemistry. Chem. Biol. Drug Des. 2015, 86, 19–65. [Google Scholar]
- Ali, A.; Muslim, M.; Kamaal, S.; Ahmed, A.; Ahmad, M.; Shahid, M.; Khan, J.A.; Dege, N.; Javed, S.; Mashrai, A. Crystal structure, Hirshfeld and electronic transition analysis of 2-[(1H-benzimidazol-1-yl)-methyl]benzoic acid. Acta Crystallogr. Sect. E 2021, 77, 755–758. [Google Scholar] [CrossRef]
- Fatima, A.; Khanum, G.; Savita, S.; Pooja, K.; Verma, I.; Siddiqui, N.; Javed, S. Quantum computational, spectroscopic, Hirshfeld surface, electronic state and molecular docking studies on sulfanilic acid: An anti-bacterial drug. J. Mol. Liq. 2021, 346, 117150. [Google Scholar] [CrossRef]
- Siddiqui, N.; Javed, S. Quantum computational, spectroscopic investigations on ampyra (4-aminopyridine) by dft/td-dft with different solvents and molecular docking studies. J. Mol. Struct. 2021, 1224, 129021. [Google Scholar] [CrossRef]
- Fatima, A.; Bhadoria, J.; Srivastava, S.K.; Verma, I.; Siddiqui, N.; Javed, S. Exploration of experimental and theoretical proper- ties of 5,5-dimethyl 3-amino-cyclohex-2-en1-one (AMINE DIMEDONE) by DFT/TD-DFT with ethanol and DMSO as solvents and molecular docking studies. J. Mol. Liq. 2021, 338, 116551. [Google Scholar] [CrossRef]
- Fatima, A.; Khanum, G.; Sharma, A.; Garima, K.; Savita, S.; Verma, I.; Siddiqui, N.; Javed, S. Computational, spectroscopic, Hirshfeld surface, electronic state and molecular docking studies on phthalic anhydride. J. Mol. Struct. 2022, 1249, 131571. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Frau, J.; Munoz, F.; Glossman-Mitnik, D. A conceptual DFT study of the chemical reactivity of magnesium octaethylporphyrin (MgOEP) as predicted by the Minnesota family of density functionals. Quím. Nova 2017, 40, 402–406. [Google Scholar] [CrossRef]
- Fatima, A.; Khanum, G.; Srivastava, S.K.; Verma, I.; Siddiqui, N.; Javed, S. Synthesis, computational, spectroscopic, hirshfeld surface, electronic state and molecular docking studies on diethyl-5-amino-3-methylthiophene-2,4-dicarboxylate. Chem. Phy. Lett. 2021, 784, 139103. [Google Scholar] [CrossRef]
- Jomroz, M.H. Vibrational Energy Distribution Analysis, VEDA4; ScienceOpen, Inc.: Warsaw, Poland, 2004. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction AnalyzerChem. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009.
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 Autodock tools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Varsanyi, G. Assignments for Vibrational Spectra of Seven Hundred Benzene Derivatives; Academic Kiado: Budapset, Hungary, 1973; Volume ½. [Google Scholar]
- Muthu, S.; Ramachandran, G.; Maheswari, J.U. Vibrational spectroscopic investigation on the structure of 2-ethylpyridine-4-carbothioamide. Spectrochim. Acta Part A 2012, 93, 214–222. [Google Scholar] [CrossRef]
- Wade, L.G. (Ed.) Advanced Organic Chemistry, 4th ed.; Wiley: New York, NY, USA, 1992; pp. 723–729. [Google Scholar]
- Jeyavijayan, S.; Arivazhagan, M. Study of density functional theory and vibrational spectra of hypoxanthine. Indian J. Pure Appl. Phys. 2010, 48, 869–874. [Google Scholar]
- Chithambarathanu, T.; Umayourbaghan, V.; Krishnakumar, V. Vibrational analysis of some pyrazole derivatives. Indian J. Pure Appl. Phys. 2003, 41, 844–848. [Google Scholar]
- Silverstein, M.; Bassler, G.C.; Morril, C. Spectroscopic Identification of Organic Compounds; John Wiley: New York, NY, USA, 1981. [Google Scholar]
- Edwin, B.; Joe, I.H. Vibrational spectra analysis of analyeuro degenerative drug Levodopa, A DFT study. J. Mol. Struct. 2013, 1034, 119–127. [Google Scholar] [CrossRef]
- Bhavani, K.; Renuga, S.; Muthu, S.; Narayanan, K.S. Quantum mechanical study and spectroscopic (FT-IR, FT-Raman, 13C, 1H) study, first order hyperpolarizability, NBO analysis, HOMO and LUMO analysis of 2-acetoxybenzoic acid by density functional methods spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 136, 1260–1268. [Google Scholar] [CrossRef]
- Ghalla, H.; Issaoui, N.; Bardak, F.; Atac, A. Intermolecular interactions and molecular docking investigations on 4-methoxybenzaldehyde. Comput. Mater. Sci. 2018, 149, 291–300. [Google Scholar] [CrossRef]
- Luque, F.J.; Lopez, J.M.; Orozco, M. Perspective on Electrostatic Interactions of a Solute with a Continuum. A Direct Utilization of AB initio Molecular Potentials for the Prevision of Solvent Effects. Theor. Chem. Acc. 2000, 103, 343–345. [Google Scholar] [CrossRef]
- Matito, E.; Sola, M. The role of electronic delocalization in transition metal complexes from the electron localization function and the quantum theory of atoms in molecules viewpoints. Coord. Chem. Rev. 2009, 253, 647–665. [Google Scholar] [CrossRef]
- Gillespie, R.J. The valence–shell electron pair repulsion (VSEPR) Theory of directed valency. J. Chem. Educ. 1963, 40, 295. [Google Scholar] [CrossRef]
- Labidi, N.S.; Djebaili, A.; Rouina, I. Substitution effects on the polarizability (α) and first hyperpolarizability (β) of all-trans hexatriene. J. Saudi Chem. Soc. 2011, 15, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, N.; Sundaraganesan, N.; Jayabharathi, J. Molecular structure, Spectroscopic (FT-IR, FT-Raman, NMR, UV) studies and first-order molecular hyperpolarizabilities of 1,2-bis(3-methoxy-4-hydroxybenzylidene)hydrazine by density functional method. Spectrochim. Acta Part A 2010, 76, 259–269. [Google Scholar] [CrossRef]
- Sarojini, K.; Krishnan, H.; Kanakam, C.C.; Muthu, S. Synthesis, structural, spectroscopic studies, NBO analysis, NLO and HOMO-LUMO of 4-methyl-N-(3-nitrophenyl)benzene sulfonamide with experimental and theoretical approaches. Spectrochim. Acta Part A 2013, 108, 159–170. [Google Scholar] [CrossRef]
- Kleinman, D.A. Non-linear Dielectric Polarization in Optical Media. Phys. Rev. 1962, 126, 1977–1979. [Google Scholar] [CrossRef]
- Khanum, G.; Fatima, A.; Siddiqui, N.; Agarwal, D.D.; Butcher, R.J.; Srivastava, S.K.; Javed, S. Synthesis, single crystal, characterization and computational study of 2-amino-N-cyclopropyl-5-ethyl-thiophene-3-carboxamide. J. Mol. Struct. 2021, 1250, 131890. [Google Scholar] [CrossRef]
- Raja, M.; Muhamed, R.R.; Muthu, S.; Suresh, M. Synthesis, spectroscopic (FT-IR, FTRaman, NMR, UV–Visible), NLO, NBO, HOMO-LUMO, Fukui function and molecular docking study of (E)-1-(5-bromo-2-hydroxybenzylidene)semicarbazide. J. Mol. Struct. 2017, 1141, 284–298. [Google Scholar] [CrossRef]
- Muthu, S.; Maheswari, J.U. Quantum mechanical study and spectroscopic (FT-IR, FT-Raman, 13C, 1H, UV) study, first order hyperpolarizability, NBO analysis, HOMO and LUMO analysis of 4-[(4-aminobenzene) sulfonyl] aniline by ab initio HF and density functional method. Spectrochim. Acta Part A 2012, 92, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Swarnalatha, N.; Gunasekaran, S.; Muthu, S.; Nagarajan, M. Molecular structure analysis and spectroscopic characterization of 9-methoxy-2H-furo[3,2-g]chromen-2-one with experimental (FT-IR and FT-Raman) techniques and quantum chemical calculations. Spectrochim. Acta Part A 2015, 137, 721–729. [Google Scholar] [CrossRef]
- Scrocco, E.; Tomasi, J. Electronic molecular structure, reactivity and intermolecular forces: Aneuristic interpretation by means of electrostatic molecular potentials. Adv. Quantum Chem. 1978, 1, 115–193. [Google Scholar]
- Raja, M.; Muhamed, R.R.; Muthu, S.; Suresh, M.; Muthu, K. Synthesis, spectroscopic (FT-IR, FT-Raman, NMR, UV–Visible), Fukui function, antimicrobial and molecular docking study of (E)-1-(3-bromobenzylidene)semicarbazide by DFT method. J. Mol. Struct. 2017, 1130, 374–384. [Google Scholar] [CrossRef]
- Vijayakumar, T.; Hubert, I.J.; Nair, C.P.R.; Jayakumar, V.S. Efficient π electrons delocalization in prospective push-pull non-linear optical chromophore 4-[N,N-dimethylamino]-4-nitrostilbene (DANS): A vibrational spectroscopic study. Chem. Phys. 2008, 343, 83–99. [Google Scholar] [CrossRef]
- Palafox, M.A. Scaling factors for the prediction of vibrational spectra of Benzene molecule. Int. J. Quantum Chem. 2000, 77, 661–684. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Rastogi, V.K.; Palafox, M.A.; Mittal, L.; Peica, N.; Keifer, W.; Lang, K.; Ojha, S.P. FTIR and FT-Raman spectra and density functional computations of the vibrational spectra, molecular geometry and atomic charges of the biomolecule:5-bromouracil. J. Raman Spectrosc. 2007, 38, 1227–1241. [Google Scholar] [CrossRef]
- Tian, L.U.; Feiwu, C. Calculation of Molecular Orbital Composition. Acta Chim. Sin. 2011, 69, 2393–2406. [Google Scholar]
- Abraham, C.S.; Muthu, S.; Prasana, J.C.; Armakovic, S.; Armakovic, S.J.; Rizwana, B.F.; Geoffrry, B.; David, A.H. Computational evaluation of the reactivity and pharmaceutical potential of an organic amine: A DFT, molecular dynamics simulations and molecular docking approach. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 222, 117–188. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond Length (Å) | Bond Angle (°) | ||||
|---|---|---|---|---|---|
| Parameter | B3LYP/6-311++G(d,p) | Experimental [19] | Parameter | B3LYP/6-311++G(d,p) | Experimental [19] |
| N22–C23 | 1.39 | 1.39(32) | C19–N17–C11 | 126.21 | 125.71(205) |
| N22–C19 | 1.30 | 1.31(32) | C11–C6–C2 | 124.04 | 124.09(224) |
| C19–N17 | 1.39 | 1.36(33) | C2–C1–O4 | 125.76 | 124.22(233) |
| N17–C20 | 1.39 | 1.39(31) | C2–C1–O3 | 112.81 | 112.24(213) |
| C20–C23 | 1.41 | 1.40(36) | C1–O3–H7 | 106.72 | 111.24(2056) |
| C20–C24 | 1.39 | 1.39(36) | O3–C1–O4 | 121.40 | 123.50(221) |
| C26–C29 | 1.40 | 1.40(37) | C1–C2–C5 | 118.41 | 117.72(224) |
| N17–C11 | 1.46 | 1.46(32) | C5–C9–C13 | 119.45 | 119.48(241) |
| C11–C6 | 1.52 | 1.50(35) | H12–C9–C13 | 120.57 | 120.26(294) |
| C6–C2 | 1.41 | 1.40(36) | Dihedral angle (°) | ||
| C2–C1 | 1.49 | 1.49(37) | C24–C20–C23–C22 | −179.98 | −179.95(228) |
| C1–O3 | 1.35 | 1.31(31) | O3–C1–C2–C5 | −3.50 | −3.54(327) |
| O3–H7 | 0.96 | 0.87(271) | O3–C1–C2–C6 | 173.75 | 173.76(233) |
| C1–O4 | 1.21 | 1.21(30) | O4–C1–C2–C5 | 178.07 | 178.07(243) |
| C2–C5 | 1.40 | 1.39(35) | O4–C1–C2–C6 | −4.66 | −4.62(403) |
| C13–C9 | 1.39 | 1.38(36) | C2–C1–O3–H7 | 178.62 | 178.26(2205) |
| C13–C10 | 1.39 | 1.39(36) | O4–C1–O3–H7 | −2.94 | −2.97(2224) |
| C10–H14 | 1.08 | 0.95(37) | C2–C6–C11–N17 | 75.19 | 75.21(309) |
| Bond angle (°) | C10–C6–C11–N17 | −105.61 | −105.61(260) | ||
| N22–C19–H21 | 125.54 | 123.15(288) | C6–C11–N17–C19 | −116.77 | −116.78(258) |
| N22–C19–N17 | 119.72 | 113.62(220) | C6–C11–N17–C20 | 64.64 | 64.66(311) |
| C19–N17–C20 | 105.86 | 106.36(199) | H15–C11–N17–C19 | 121.9 | 121.85(298) |
| C20–C23–N22 | 110.22 | 109.22(216) | H15–C11–N17–C20 | −56.68 | −56.69(360) |
| C23–C25–H28 | 120.14 | 121.25(298) | H16–C11–N17–C19 | 4.60 | 4.610(377) |
| C23–C25–C29 | 118.12 | 117.52(239) | H16–C11–N17–C20 | −173.97 | −173.93(268) |
| C25–C29–H31 | 119.64 | 119.45(304) | C11–N17–C19–N22 | −179.61 | −179.62(215) |
| C29–C26–C24 | 121.56 | 122.13(236) | C20–N17–C19–N22 | −0.78 | −0.82(285) |
| C24–C20–N17 | 133.05 | 133.14(228) | N17–C19–N22–C23 | 0.28 | 0.28(284) |
| C20–N17–C11 | 127.64 | 127.89(198) | H21–C19–N22–C23 | −179.70 | −179.73(313) |
| N17–C11–C6 | 114.46 | 113.31(206) | H21–C19–C23–N22 | −0.8 | −0.84(271) |
| Parameters | B3LYP/6-311++G(d,p) | Urea [49] | B3LYP/6-311++G(d,p) | Parameters | B3LYP/6-311++G(d,p) | Urea [49] | B3LYP/6-311++G(d,p) |
|---|---|---|---|---|---|---|---|
| μx | −4.28 | −0.806 | 0.9450 | βxxx | −44.37 | 23.748 | 0.9595 |
| μy | 3.12 | 1.543 | −1.1264 | βyxx | 17.74 | 17.376 | −1.4268 |
| μz | 0.35 | −0.008 | 0.0000 | βxyy | −48.53 | −55.468 | −1.4018 |
| μ(D) | 5.31 | −1.741 | 1.4703 | βyyy | 6.84 | 44.220 | −3.3043 |
| αxx | −110.09 | 37.245 | −25.0247 | βzxx | −42.35 | −0.489 | 0.0000 |
| αxy | 4.37 | −0.194 | 0.6994 | βxyz | −9.41 | 0.034 | 0.0000 |
| αyy | −104.73 | 37.988 | −23.1428 | βzyy | −13.38 | −0.531 | 0.0000 |
| αxz | 2.194 | 0.052 | 0.0000 | βxzz | −11.73 | −19.037 | 3.6839 |
| αyz | 3.826 | −0.063 | 0.0000 | βyzz | −12.69 | 33.038 | −6.5824 |
| αzz | −107.97 | 24.012 | −6.7450 | βzzz | 8.39 | −1.062 | 0.0000 |
| α0 (e.s.u) | −1.594 × 510−23 | 0.9771 × 10−23 | 0.897 × 10−23 | βtot (e.s.u) | 0.9975 × 10−31 | 0.927 × 10−30 | 0.917 × 10−31 |
| Atom | Mulliken Atomic Charges | Fukui Functions | Local Softness | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| N (0,1) | N-1 (+1,2) | N+1(-1,2) | fr+ | fr- | Δf | fr0 | sr+ fr+ | sr- fr- | sr0 fr0 | |
| C1 | −0.245 | −0.259 | −0.211 | 0.034 | 0.014 | 0.020 | −0.081 | 0.008 | 0.003 | −0.020 |
| C2 | 0.386 | 0.397 | 0.295 | −0.090 | −0.011 | −0.078 | 0.096 | −0.022 | −0.002 | 0.023 |
| O3 | −0.128 | −0.114 | −0.183 | −0.055 | −0.014 | −0.040 | −0.126 | −0.013 | −0.003 | −0.031 |
| O4 | −0.254 | −0.249 | −0.399 | −0.144 | −0.004 | −0.140 | −0.274 | −0.035 | −0.001 | −0.067 |
| C5 | −0.254 | 0.238 | −0.327 | −0.073 | −0.493 | 0.420 | −0.446 | −0.018 | −0.121 | −0.110 |
| C6 | 0.934 | 0.890 | 0.977 | 0.043 | 0.043 | −9.9E- | 0.5325 | 0.010 | 0.010 | 0.131 |
| C9 | −0.531 | −0.503 | −0.553 | −0.022 | −0.027 | 0.004 | −0.301 | −0.005 | −0.006 | −0.074 |
| C10 | −0.271 | −0.239 | −0.293 | −0.021 | −0.031 | 0.009 | −0.173 | −0.005 | −0.007 | −0.042 |
| C11 | −0.610 | −0.691 | −0.694 | −0.084 | 0.080 | −0.165 | −0.349 | −0.020 | 0.019 | −0.086 |
| C13 | 0.342 | −0.335 | −0.462 | −0.805 | 0.678 | −1.483 | −0.294 | −0.198 | 0.167 | −0.072 |
| N17 | 0.198 | 0.280 | 0.213 | 0.014 | −0.082 | 0.097 | 0.072 | 0.003 | −0.020 | 0.017 |
| C19 | 0.129 | 0.147 | 0.123 | −0.006 | −0.018 | 0.012 | 0.0490 | −0.001 | −0.004 | 0.012 |
| C20 | 0.179 | 0.188 | 0.219 | 0.040 | −0.009 | 0.049 | 0.1254 | 0.009 | −0.002 | 0.030 |
| N22 | −0.072 | 0.042 | −0.101 | −0.028 | −0.115 | 0.086 | −0.122 | −0.007 | −0.028 | −0.030 |
| C23 | −0.105 | −0.116 | −0.107 | −0.002 | 0.010 | −0.013 | −0.049 | −0.000 | 0.002 | −0.012 |
| C24 | −0.327 | −0.228 | −0.333 | −0.006 | −0.099 | 0.093 | −0.219 | −0.001 | −0.024 | −0.054 |
| C25 | −0.379 | −0.294 | −0.391 | −0.011 | −0.085 | 0.073 | −0.243 | −0.002 | −0.021 | −0.060 |
| C26 | −0.262 | −0.248 | −0.301 | −0.039 | −0.013 | −0.026 | −0.176 | −0.009 | −0.003 | −0.043 |
| C29 | −0.308 | −0.253 | −0.303 | 0.005 | −0.055 | 0.061 | −0.176 | 0.001 | −0.013 | −0.043 |
| Parameter | Values |
|---|---|
| EHomo(eV) | −6.20170 |
| ELumo(eV) | −2.14768 |
| Ionization potential | 6.20170 |
| Electron affinity | 2.14768 |
| Energy gap(eV) | 4.0540 |
| Electronegativity | 4.1746 |
| Chemical potential | −4.1746 |
| Chemical hardness | 2.0270 |
| Chemical softness | 0.2466 |
| Electrophilicity index | 4.2987 |
| Experimental | TD/DFT | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Solvent | λmax (nm) | Molar Extinction Coefficient | Band Energy (eV) | Energy | λmax (nm) | Band Energy (eV) | Energy | Oscillator Strength | Assignments |
| Gas | 357 | 3.47 | 28,021.3 | 0.0002 | H→L (96.6%) | ||||
| 347 | 3.57 | 28,816.56 | 0.0001 | H-1→L (96.7%) | |||||
| 279 | 4.43 | 35,792.45 | 0.0183 | H→L+1 (91.2%) | |||||
| Ethanol | 271.92 | 271,920 | 4.56 | 36,779 | 311 | 3.98 | 32,133.11 | 0.0006 | H→L (93.3%) |
| 304 | 4.08 | 32,934.83 | 0.0002 | H-1→L (93.7%) | |||||
| 261 | 4.74 | 38,288.73 | 0.0494 | H-2→L (72.3%) | |||||
| DCM | 352.03 | 352,029.9 | 3.52 | 28,391 | 315 | 3.93 | 31,700.8 | 0.0006 | H→L (93.1%) |
| 308 | 4.02 | 32,468.64 | 0.0002 | H-1→L (93.4%) | |||||
| 261 | 4.74 | 38,294.38 | 0.0493 | H-2→L (70.9%) | |||||
| DMSO | 396.42 | 396,420 | 3.13 | 25,245 | 310 | 3.99 | 32,245.22 | 0.0007 | H→L (93.5%) |
| 302 | 4.085 | 33,056.62 | 0.0003 | H-1→L (93.8%) | |||||
| 261 | 4.74 | 38,258.08 | 0.0547 | H-2→L (72.4%) | |||||
| Atoms | Experimental Chemical Shift (ppm) | Calculated Chemical Shift (ppm) B3LYP | Degeneracy (ppm) | RMSD (R) and R2 Values |
|---|---|---|---|---|
| 8H | 8.25 | 8.577 | 1.000 | For 1H |
| 21H | 7.88 | 8.350 | 2.000 | R = 0.956 |
| 14H | 7.86 | 8.304 | 2.000 | R2 = 0.914 |
| 18H | 7.36 | 8.180 | 1.000 | |
| 28H | 7.34 | 8.031 | 1.000 | |
| 12H | 7.32 | 7.911 | 1.000 | |
| 27H | 7.15 | 7.659 | 1.000 | |
| 7H | 7.14 | 7.587 | 3.000 | |
| 31H | 7.13 | 7.575 | 3.000 | |
| 30H | 7.12 | 7.536 | 3.000 | |
| 16H | 6.72 | 6.819 | 1.000 | |
| 15H | 5.82 | 5.216 | 1.000 |
| Centroid Coordinates | |||||||
|---|---|---|---|---|---|---|---|
| Solvent | Excited State | λmax | f | Maps | X(Å) | Y(Å) | Z(Å) |
| DCM | 3 | 261.13 | 0.0493 | EDD | −2.16 | 0.24 | 0.01 |
| HDD | −2.16 | 0.94 | 0.08 | ||||
| Distance between Centroid | 0.00 | 0.70 | 0.06 | ||||
| EtOH | 3 | 261.17 | 0.0494 | EDD | −2.16 | 0.23 | 0.12 |
| HDD | −2.16 | 0.96 | 0.07 | ||||
| Distance between Centroid | 0.00 | 0.72 | 0.06 | ||||
| S.No. | Derivatives | HBD | HBA | MR | TPSA (Å2) | GI Absorption | BBB Permanent | CYP1A2 Inhibitor | log Kp (cm/s) | Lipinski Violations | Bioavailability Score |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 21HBMBA | 1 | 3 | 72.44 | 55.12 | High | Yes | Yes | −6.02 | Yes | 0.85 |
| 2 | 2-[4-[(2-propylben-zo[f]benzimidazol-3-yl)methyl]-phenyl]benzoic acid | 1 | 3 | 129.96 | 55.12 | High | Yes | Yes | −4.42 | Yes | 0.85 |
| 3 | 1-cyclohexyl-2-phenylbenzimidazole-5-carboxylic acid | 1 | 3 | 95.31 | 55.12 | High | Yes | Yes | −5.17 | Yes | 0.85 |
| 4 | 2-[4-[(2-choloro-ethyl)benzimidazol-1-yl)methyl]-phenyl]-benzoic acid | 1 | 3 | 130.01 | 55.12 | High | Yes | No | −4.22 | Yes | 0.85 |
| S. No. | PDB ID | Residue | Bond Distance (Å) | Inhibition Constant (Micromolar) | Binding Energy (kcal/mol) | Reference RMSD (Å) |
|---|---|---|---|---|---|---|
| 1 | 3H4G | 3 | 1.930,2.366 | 0.17 | −9.2 | 10.746 |
| 2 | 4A7Y | 3 | 2.412 | 0.48 | −8.6 | 21.907 |
| 3 | 6M7X | 3 | 2.259,2.383 | 1.87 | −7.8 | 9.774 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khanum, G.; Ali, A.; Shabbir, S.; Fatima, A.; Alsaiari, N.; Fatima, Y.; Ahmad, M.; Siddiqui, N.; Javed, S.; Gupta, M. Vibrational Spectroscopy, Quantum Computational and Molecular Docking Studies on 2-[(1H-Benzimidazol-1-yl)-methyl]benzoic Acid. Crystals 2022, 12, 337. https://doi.org/10.3390/cryst12030337
Khanum G, Ali A, Shabbir S, Fatima A, Alsaiari N, Fatima Y, Ahmad M, Siddiqui N, Javed S, Gupta M. Vibrational Spectroscopy, Quantum Computational and Molecular Docking Studies on 2-[(1H-Benzimidazol-1-yl)-methyl]benzoic Acid. Crystals. 2022; 12(3):337. https://doi.org/10.3390/cryst12030337
Chicago/Turabian StyleKhanum, Ghazala, Arif Ali, Sadiya Shabbir, Aysha Fatima, Norah Alsaiari, Yasmeen Fatima, Musheer Ahmad, Nazia Siddiqui, Saleem Javed, and Mayank Gupta. 2022. "Vibrational Spectroscopy, Quantum Computational and Molecular Docking Studies on 2-[(1H-Benzimidazol-1-yl)-methyl]benzoic Acid" Crystals 12, no. 3: 337. https://doi.org/10.3390/cryst12030337
APA StyleKhanum, G., Ali, A., Shabbir, S., Fatima, A., Alsaiari, N., Fatima, Y., Ahmad, M., Siddiqui, N., Javed, S., & Gupta, M. (2022). Vibrational Spectroscopy, Quantum Computational and Molecular Docking Studies on 2-[(1H-Benzimidazol-1-yl)-methyl]benzoic Acid. Crystals, 12(3), 337. https://doi.org/10.3390/cryst12030337