
Revision of the Crystal Structure of the Orthorhombic Polymorph of Oxyma: On the Importance of π–Hole Interactions and Their Interplay with H–Bonds

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials, Experimental and Theoretical Methods
2.1. Materials
2.2. Synthesis of the Single Crystals
2.3. Single Crystal X-ray Diffraction
2.4. Computational Details
3. Results and Discussion
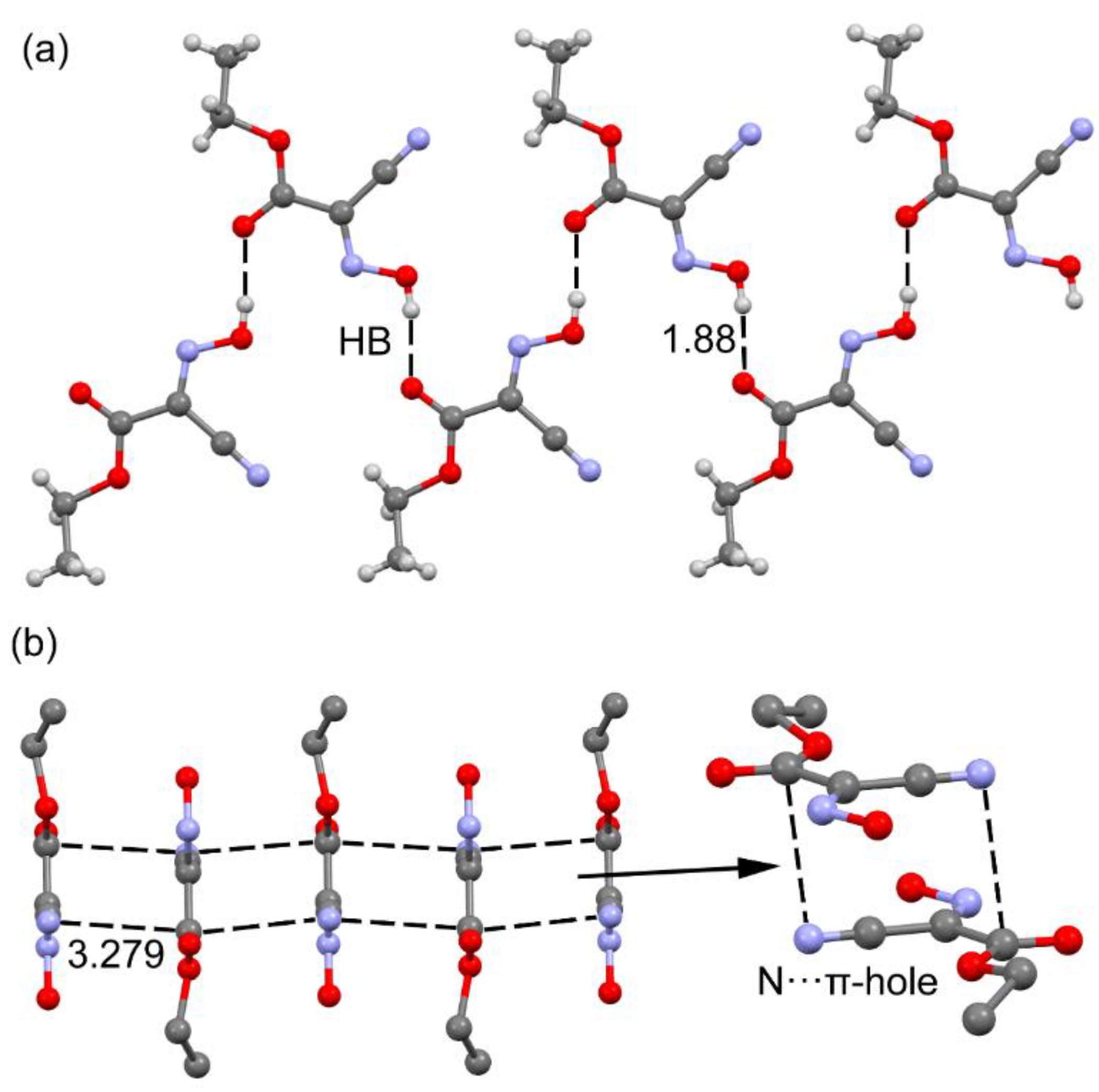
3.1. Revision of the Crystal Structure of the Orthorhombic Polymorph of Oxyma
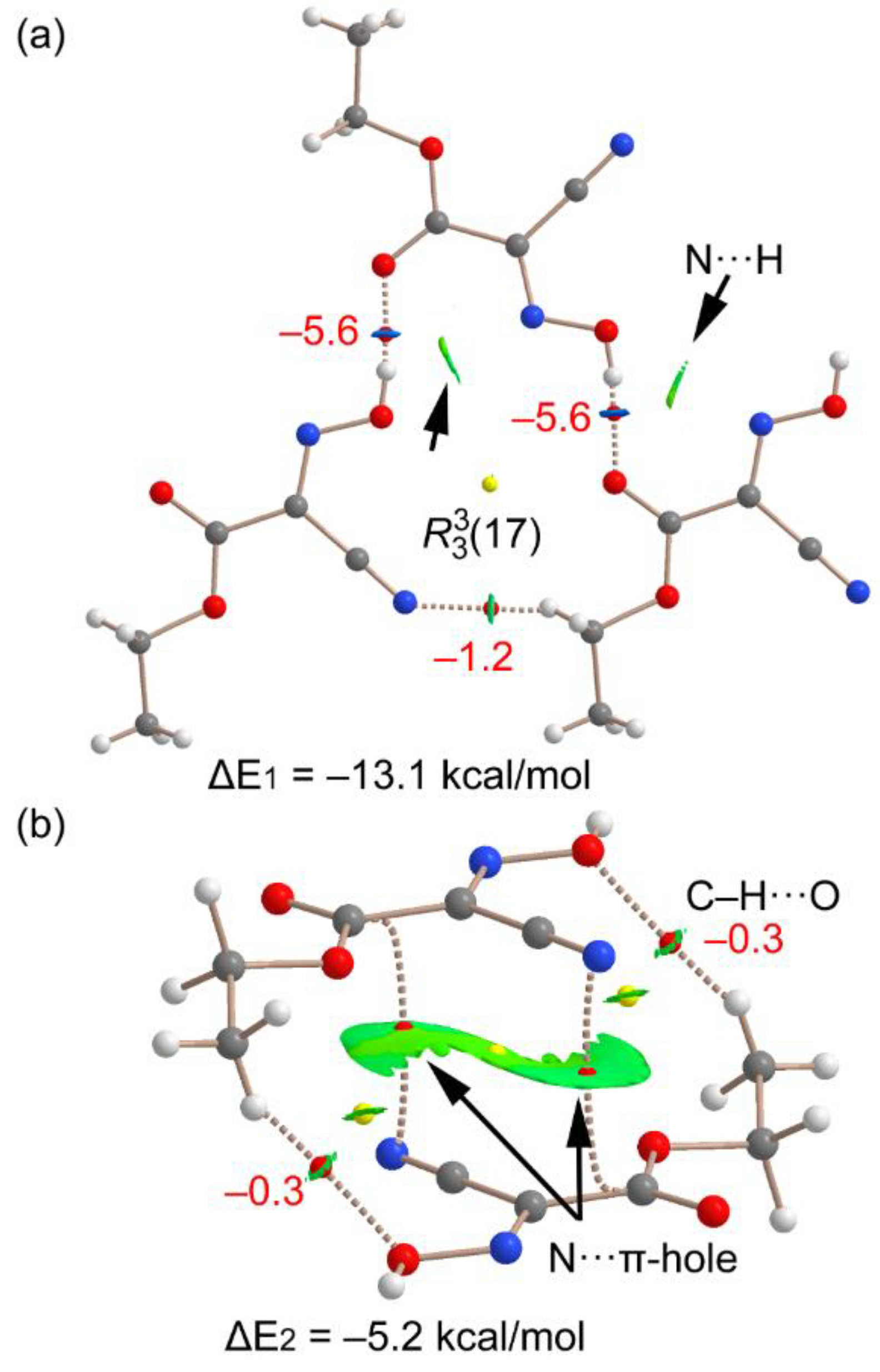
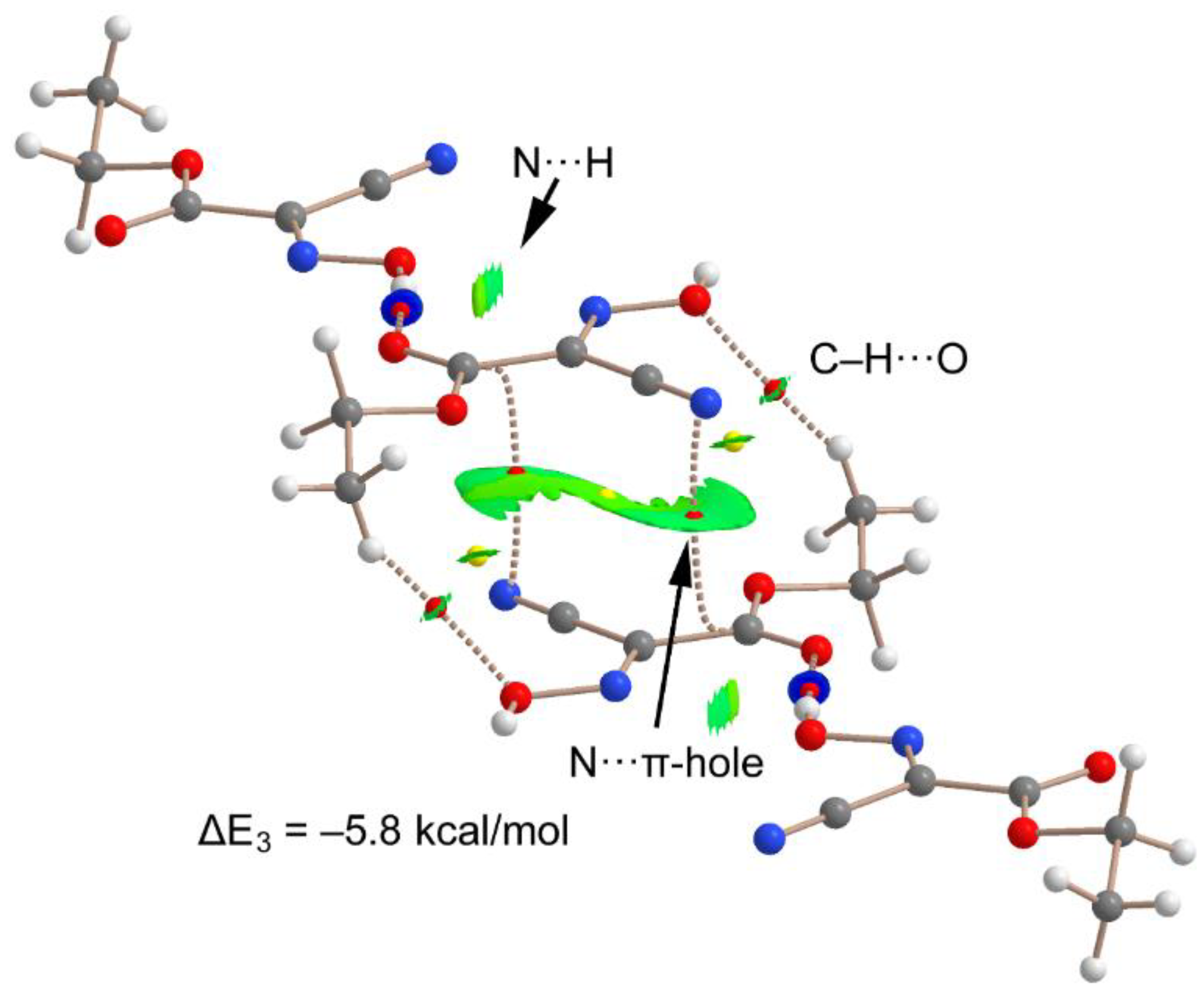
3.2. DFT Calculations
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pimentel, G.C.; McClellan, A.L. The Hydrogen Bond; W.H. Freeman & Co.: San Francisco, CA, USA, 1960. [Google Scholar]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond. In Structural Chemistry and Biology; International Union of Crystallography, Oxford Science Publications: Oxford, UK, 1999. [Google Scholar]
- Gilli, G.; Gilli, P. The Nature of the Hydrogen Bond: Outline of a Comprehensive Hydrogen Bond Theory; International Union of Crystallography, Oxford Science Publications: Oxford, UK, 2009. [Google Scholar]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. The bright future of unconventional σ/π-hole interactions. ChemPhysChem 2015, 16, 2496–2517. [Google Scholar] [CrossRef] [PubMed]
- Angarov, V.; Kozuch, S. On the σ, π and δ hole interactions: A molecular orbital overview. New J. Chem. 2018, 42, 1413–1422. [Google Scholar] [CrossRef]
- Gao, L.; Zeng, Y.; Zhang, X.; Meng, L. Comparative studies on group III σ-hole and π-hole interactions. J. Comput. Chem. 2016, 37, 1321–1327. [Google Scholar] [CrossRef]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. Directionality of π-holes in nitro compounds. Chem. Commun. 2015, 51, 1491–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, J.S.; Lane, P.; Clark, T.; Riley, K.E.; Politzer, P. σ-Holes, π-holes and electrostatically-driven interactions. J. Mol. Model. 2012, 18, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Noncovalent Bonds through Sigma and Pi-Hole Located on the Same Molecule. Guiding Principles and Comparisons. Molecules 2021, 26, 1740. [Google Scholar] [CrossRef]
- Burgi, H.B.; Dunitz, J.D.; Shefter, E. Geometrical reaction coordinates. II. Nucleophilic addition to a carbonyl group. J. Am. Chem. Soc. 1973, 95, 5065–5067. [Google Scholar] [CrossRef]
- Harder, M.; Kuhn, B.; Diederich, F. Efficient Stacking on Protein Amide Fragments. Chemmedchem 2013, 8, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, G.J.; Choudhary, A.; Raines, R.T.; Woolfson, D.N. n→π* interactions in proteins. Nat. Chem. Biol. 2010, 6, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Luxemburg Bio Technologies. Available online: https://www.luxembourg-bio.com/products/OxymaPure (accessed on 12 April 2022).
- Subirós-Funosas, R.; Prohens, R.; Barbas, R.; El-Faham, A.; Albericio, F. Oxyma: An Efficient Additive for Peptide Synthesis to Replace the Benzotriazole-Based HOBt and HOAt with a Lower Risk of Explosion. Chem. Eur. J. 2009, 15, 9394–9403. [Google Scholar] [CrossRef] [PubMed]
- Wehrstedt, K.D.; Wandrey, P.A.; Heitkamp, D. Explosive properties of 1-hydroxybenzotriazoles. J. Hazard. Mater. 2005, A126, 1–7. [Google Scholar] [CrossRef] [PubMed]
- El-Faham, A.; Al Marhoon, Z.; Abdel-Megeed, A.; Albericio, F. OxymaPure/DIC: An Efficient Reagent for the Synthesis of a Novel Series of 4-[2-(2-Acetylaminophenyl)-2-oxo-acetylamino] Benzoyl Amino Acid Ester Derivatives. Molecules 2013, 18, 14747–14759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opalade, A.A.; Gomez-Garcia, C.J.; Gerasimchuk, N. New Route to Polynuclear Ni(II) and Cu(II) Complexes with Bridging Oxime Groups That Are Inaccessible by Conventional Preparations. Cryst. Growth Des. 2019, 19, 678–693. [Google Scholar] [CrossRef]
- SADABS Bruker AXS.; Madison, Wisconsin, USA, 2004; SAINT, Software Users Guide, Version 6.0; Bruker Analytical X-ray Systems: Madison, WI, 1999. Sheldrick, G.M. SADABS v2.03; Area-Detector Absorption Correction; University of Göttingen: Germany, 1999. Saint, Version 7.60A.; Bruker AXS 2008; SADABS, V. 2008-1, 2008.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154118. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. J. Mol. Phys. 1970, 19, 553–566. [Google Scholar]
- Bader, R.F.W. A Bond Path: A Universal Indicator of Bonded Interactions. J. Phys. Chem. A 1998, 102, 7314–7323. [Google Scholar] [CrossRef]
- Keith, T.A. TK Gristmill Software, version 13.05.06; AIMAll: Overland Park, KS, USA, 2013.
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W.J. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W.J. Revealing noncovalent interactions. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Minacheva, L.K.; Raspertova, I.V.; Sliva, T.Y.; Lampeka, R.D. Crystal and Molecular Structures of 2-Cyano-2-Hydroxyiminoacetic Acid Ethyl Ester, HONC(CN)C(O)C2H5. Russ. J. Coord. Chem. 2000, 26, 226–228. [Google Scholar]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring Nature and Predicting Strength of Hydrogen Bonds: A Correlation Analysis Between Atoms-in-Molecules Descriptors, Binding Energies, and Energy Components of Symmetry-Adapted Perturbation Theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbas, R.; de Sande, D.; Font-Bardia, M.; Prohens, R.; Frontera, A. Revision of the Crystal Structure of the Orthorhombic Polymorph of Oxyma: On the Importance of π–Hole Interactions and Their Interplay with H–Bonds. Crystals 2022, 12, 823. https://doi.org/10.3390/cryst12060823
Barbas R, de Sande D, Font-Bardia M, Prohens R, Frontera A. Revision of the Crystal Structure of the Orthorhombic Polymorph of Oxyma: On the Importance of π–Hole Interactions and Their Interplay with H–Bonds. Crystals. 2022; 12(6):823. https://doi.org/10.3390/cryst12060823
Chicago/Turabian StyleBarbas, Rafael, Dafne de Sande, Mercè Font-Bardia, Rafel Prohens, and Antonio Frontera. 2022. "Revision of the Crystal Structure of the Orthorhombic Polymorph of Oxyma: On the Importance of π–Hole Interactions and Their Interplay with H–Bonds" Crystals 12, no. 6: 823. https://doi.org/10.3390/cryst12060823
APA StyleBarbas, R., de Sande, D., Font-Bardia, M., Prohens, R., & Frontera, A. (2022). Revision of the Crystal Structure of the Orthorhombic Polymorph of Oxyma: On the Importance of π–Hole Interactions and Their Interplay with H–Bonds. Crystals, 12(6), 823. https://doi.org/10.3390/cryst12060823