
Bifurcated Halogen Bond-Driven Supramolecular Double Helices from 1,2-Dihalotetrafluorobenzene and 2,2′-Bi(1,8-naphthyridine)


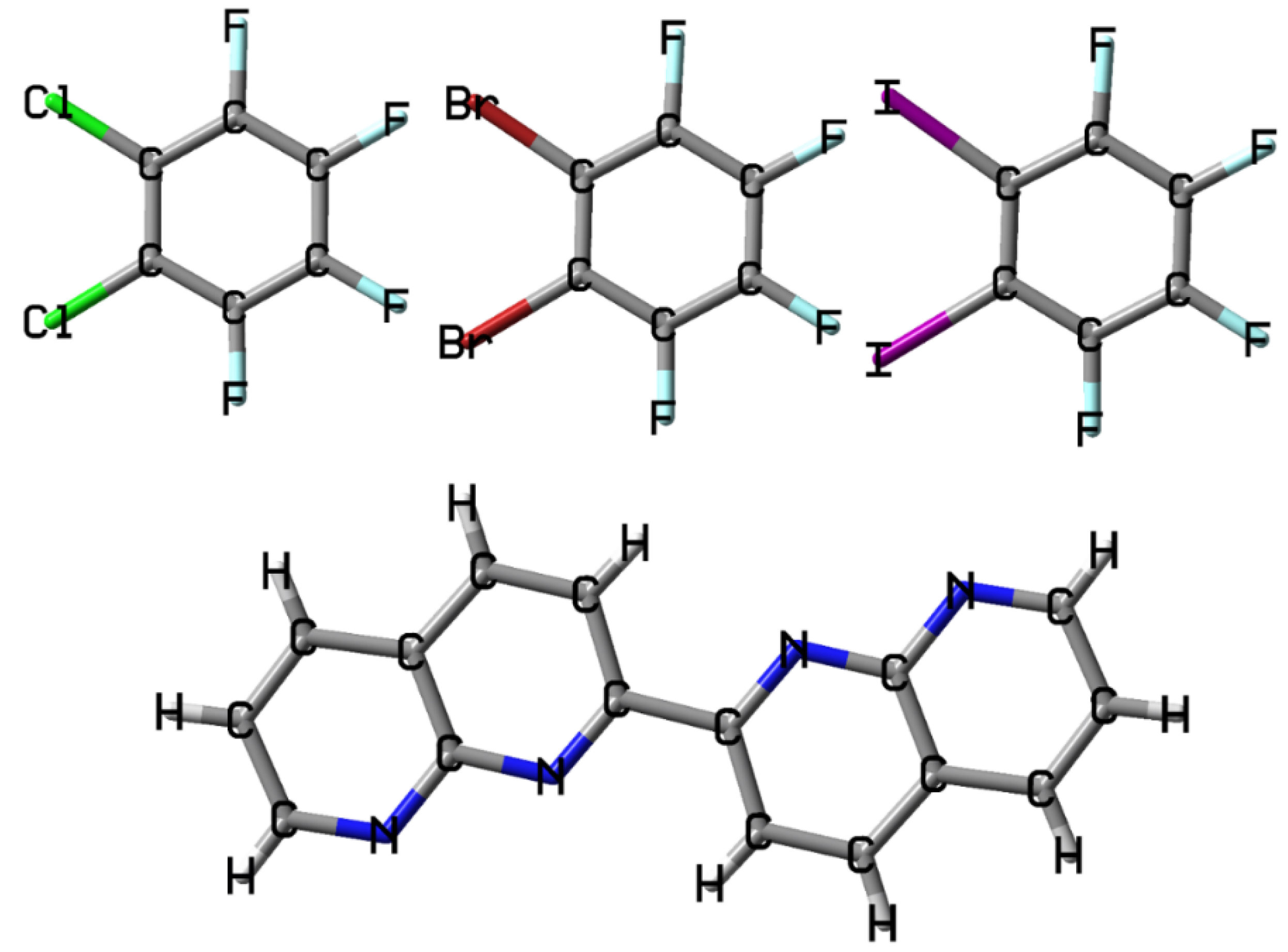{kind=link}
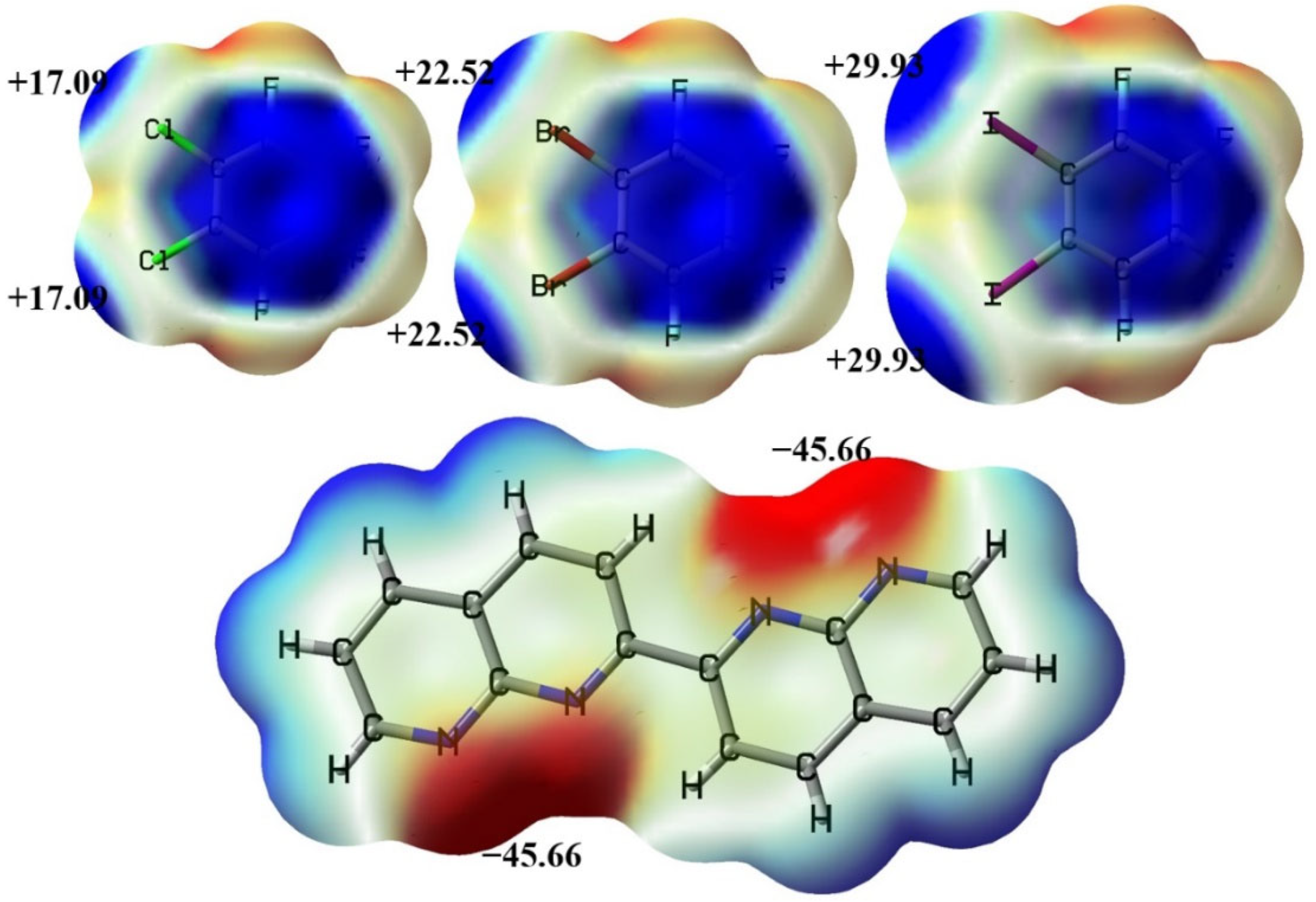{kind=link}
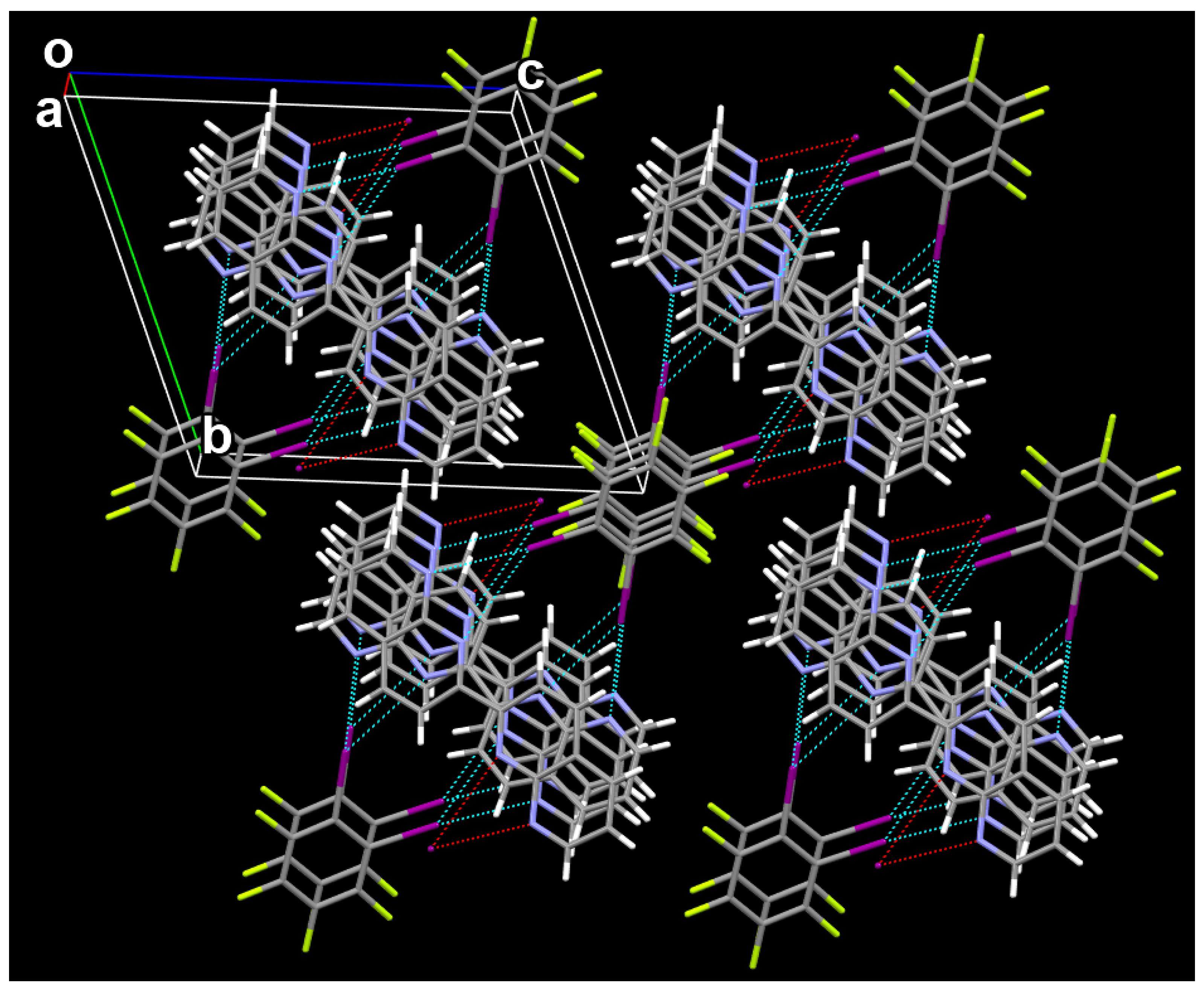{kind=link}
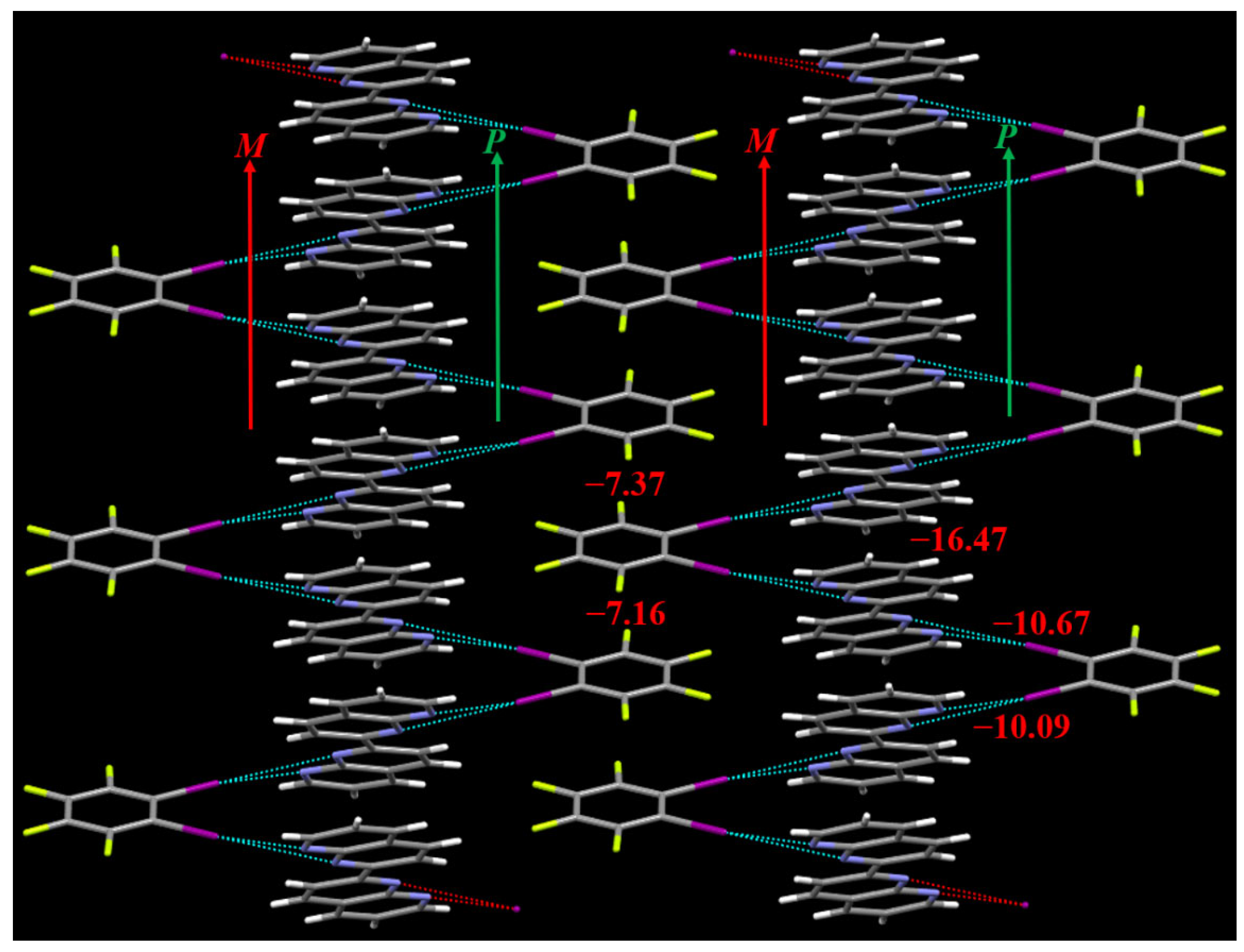{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cocrystal Synthesis
2.2. X-ray Crystallography
2.3. Computational Details
3. Results and Discussion
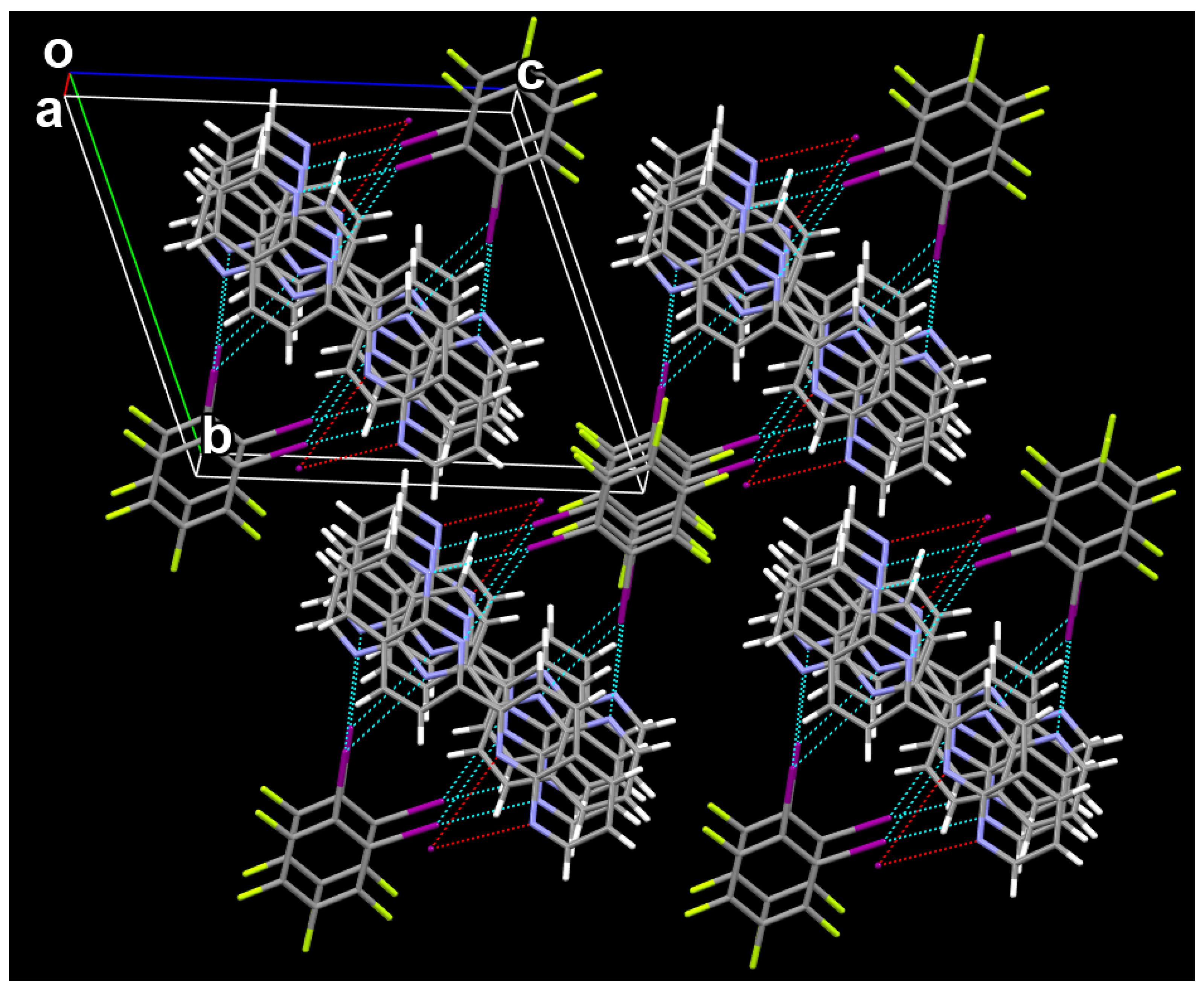
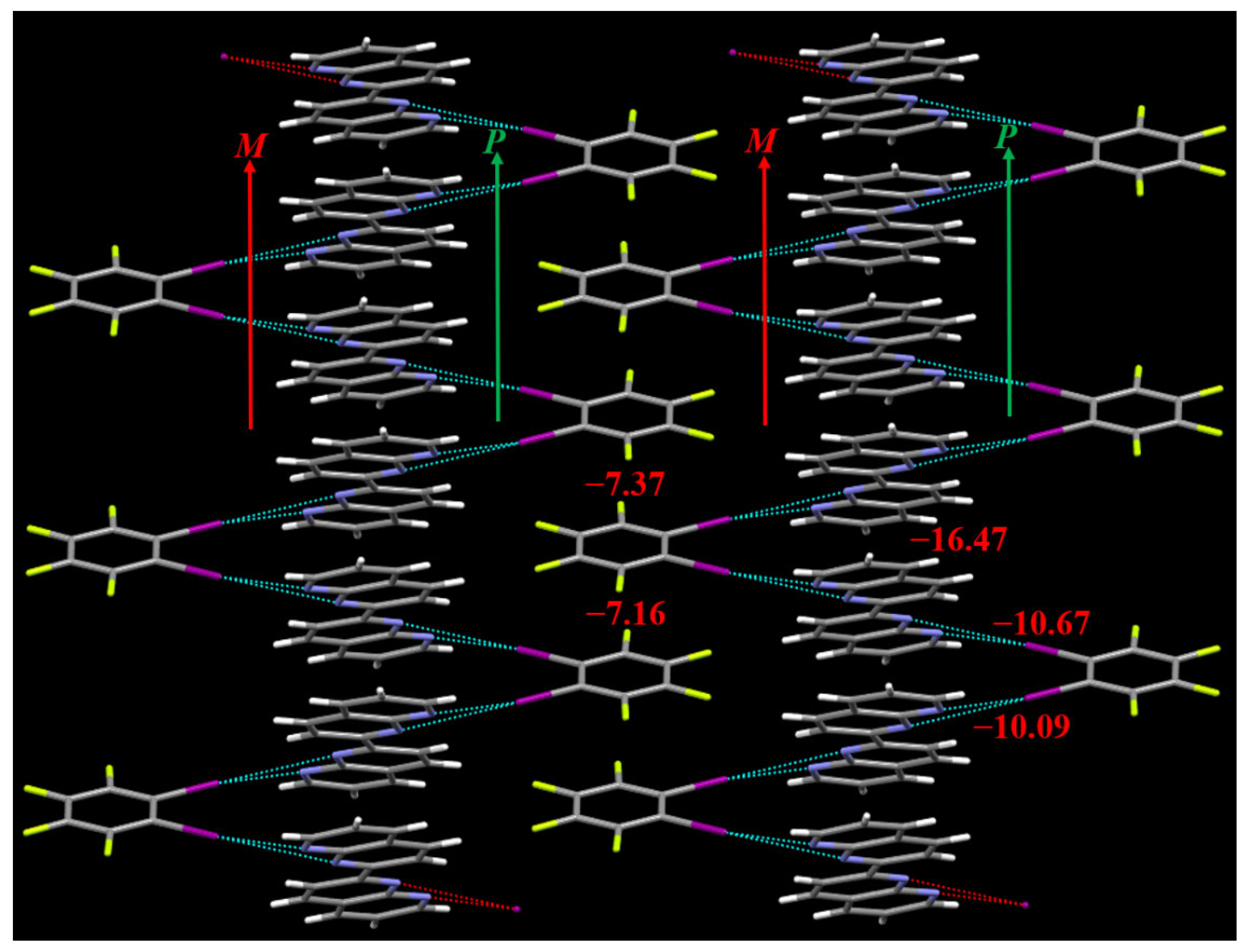
3.1. The Cocrystal Structure
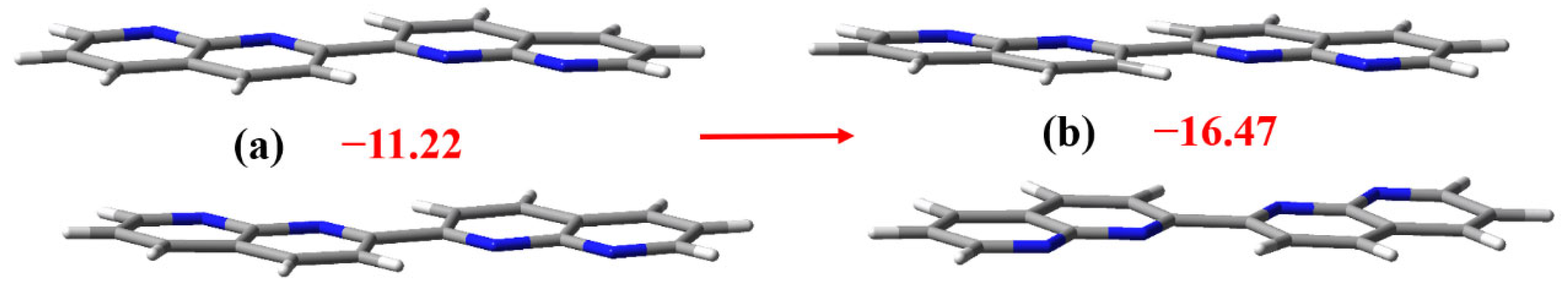
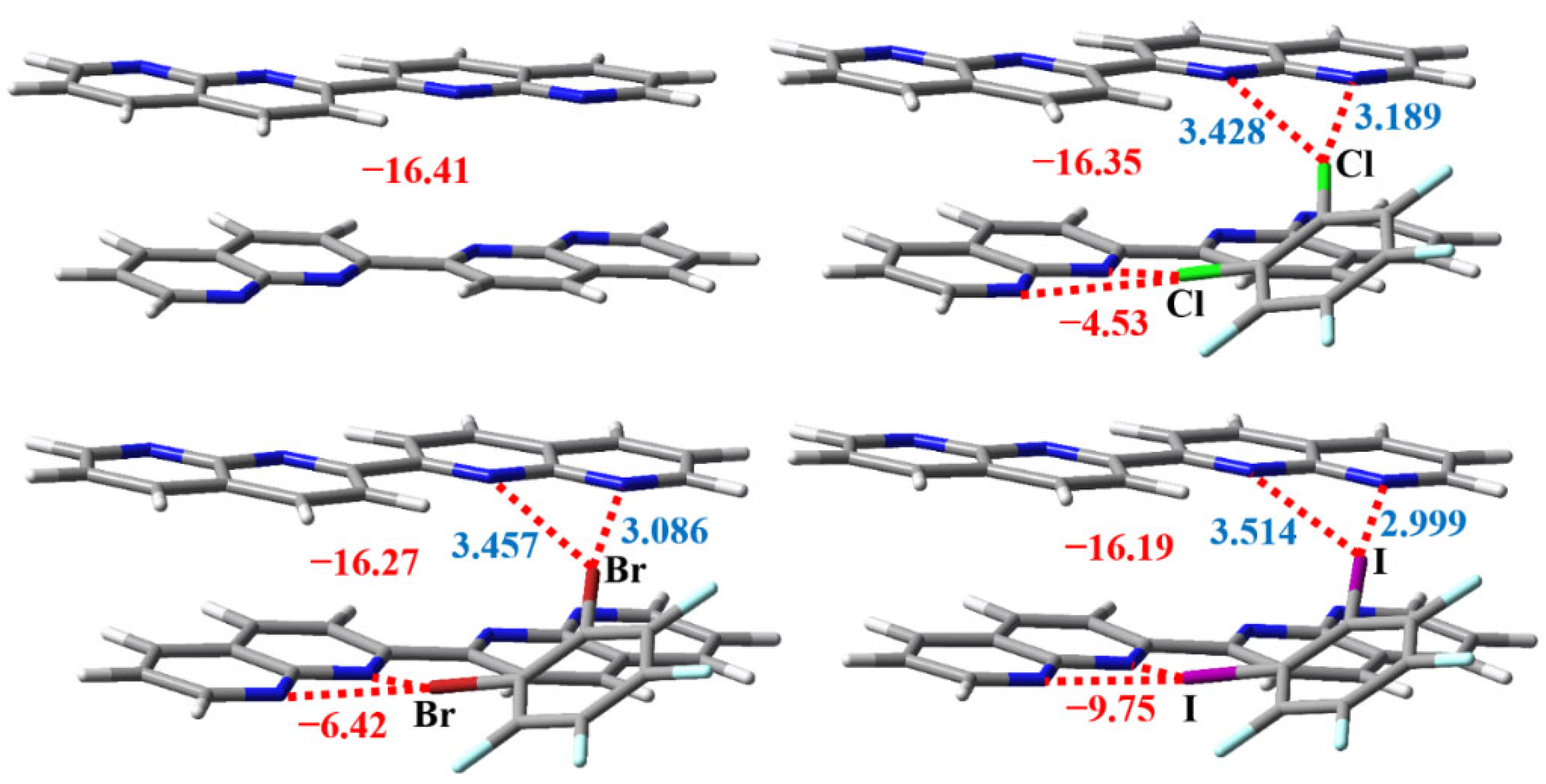
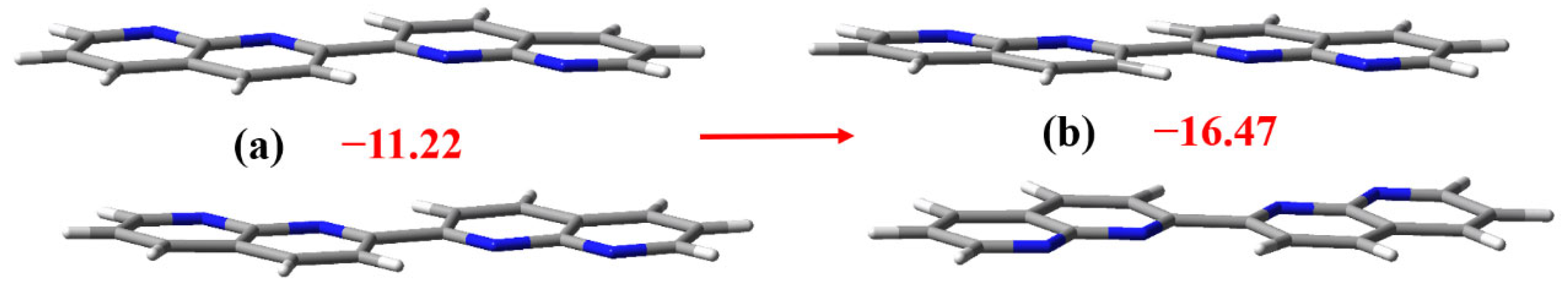
3.2. The Noncovalent Interactions in the Gas Phase
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, W.; Jin, W.J. σ-Hole Bond vs π-Hole Bond: A Comparison Based on Halogen Bond. Chem. Rev. 2016, 116, 5072–5104. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, Y.; Jin, W.J. Halogen Bonding in Room-Temperature Phosphorescent Materials. Coord. Chem. Rev. 2020, 404, 213107. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Frontera, A. Not Only Hydrogen Bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef] [Green Version]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen Bonding: The σ-Hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Bulatova, M.; Ivanov, D.M.; Haukka, M. Classics Meet Classics: Theoretical and Experimental Studies of Halogen Bonding in Adducts of Platinum(II) 1,5-Cyclooctadiene Halide Complexes with Diiodine, Iodoform, and 1,4-Diiodotetrafluorobenzene. Cryst. Growth Des. 2021, 21, 974–987. [Google Scholar] [CrossRef]
- Rajakumar, K.; Sharutin, V.V.; Adonin, S.A.; Zherebtsov, D.A.; Sakhapov, I.F.; Islamov, D.R.; Prabunatan, P.; Vershinin, M.A.; Naifert, S.A.; Polozov, M.A. Di- and Tetraiodoxylenes: Structure and Features of Non-Covalent Interaction in a Solid State. J. Struct. Chem. 2022, 63, 620–625. [Google Scholar] [CrossRef]
- Kryukova, M.A.; Sapegin, A.V.; Novikov, A.S.; Krasavin, M.; Ivanov, D.M. New Crystal Forms for Biologically Active Compounds. Part 2: Anastrozole as N-Substituted 1,2,4-Triazole in Halogen Bonding and Lp-π Interactions with 1,4-Diiodotetrafluorobenzene. Crystals 2020, 10, 371. [Google Scholar] [CrossRef]
- Eliseeva, A.A.; Ivanov, D.M.; Novikov, A.S.; Kukushkin, V.Y. Recognition of the π-Hole Donor Ability of Iodopentafluorobenzene—a Conventional σ-Hole Donor for Crystal Engineering Involving Halogen Bonding. CrystEngComm 2019, 21, 616–628. [Google Scholar] [CrossRef]
- Ragusa, A.C.; Peloquin, A.J.; McMillen, C.D.; Pennington, W.T. 2,5-Diiodothiophene: A Versatile Halogen Bonding Synthon for Crystal Engineering. Cryst. Growth Des. 2022, 22, 1906–1913. [Google Scholar] [CrossRef]
- Bondarenko, M.A.; Abramov, P.A.; Novikov, A.S.; Sokolov, M.N.; Adonin, S.A. Cu(II) Pentaiodobenzoate Complexes: “Super Heavy Carboxylates” Featuring Strong Halogen Bonding. Polyhedron 2022, 214, 115644. [Google Scholar] [CrossRef]
- Posavec, L.; Nemec, V.; Stilinović, V.; Cinčić, D. Halogen and Hydrogen Bond Motifs in Ionic Cocrystals Derived from 3-Halopyridinium Halogenides and Perfluorinated Iodobenzenes. Cryst. Growth Des. 2021, 21, 6044–6050. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Han, J.; Jin, X.; Ye, Q.; Zhou, J.; Duan, P.; Liu, M. Halogen Bonded Chiral Emitters: Generation of Chiral Fractal Architecture with Amplified Circularly Polarized Luminescence. Angew. Chem. Int. Ed. 2021, 60, 22711–22716. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Comparison of Bifurcated Halogen with Hydrogen Bonds. Molecules 2021, 26, 350. [Google Scholar] [CrossRef]
- Cinčić, D.; Friščić, T.; Jones, W. Experimental and Database Studies of Three-Centered Halogen Bonds with Bifurcated Acceptors Present in Molecular Crystals, Cocrystals and Salts. CrystEngComm 2011, 13, 3224–3231. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Wijethunga, T.K.; Desper, J. Constructing Molecular Polygons Using Halogen Bonding and Bifurcated N-Oxides. CrystEngComm 2014, 16, 28–31. [Google Scholar] [CrossRef] [Green Version]
- Efimenko, Z.M.; Eliseeva, A.A.; Ivanov, D.M.; Galmés, B.; Frontera, A.; Bokach, N.A.; Kukushkin, V.Y. Bifurcated μ2-I···(N,O) Halogen Bonding: The Case of (Nitrosoguanidinate)NiII Cocrystals with Iodine(I)-Based σ-Hole Donors. Cryst. Growth Des. 2021, 21, 588–596. [Google Scholar] [CrossRef]
- Gao, Y.J.; Li, C.; Liu, R.; Jin, W.J. Phosphorescence of Several Cocrystals Assembled by Diiodotetrafluorobenzene and Three Ring Angular Diazaphenanthrenes via C–I···N Halogen Bond. Spectrochim. Acta Part A 2017, 173, 792–799. [Google Scholar] [CrossRef]
- Sadhukhan, N.; Saha, S.; Bera, J.K. Multinuclear Complexes Derived from Bi-1, 8-naphthyridine Ligands. J. Indian Chem. Soc. 2015, 92, 1957–1964. [Google Scholar]
- Cavallo, G.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Naming Interactions from the Electrophilic Site. Cryst. Growth Des. 2014, 14, 2697–2702. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen Bonding: An Electrostatically-Driven Highly Directional Noncovalent Interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef] [PubMed]
- CrysAlisPro, Rigaku Oxford Diffraction, version 1.171.39.46; Oxford Diffraction Ltd.: Yarnton, UK, 2018.
- Sheldrick, G.M. SHELXT–Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Difference of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Wang, W.; Sun, T.; Zhang, Y.; Wang, Y.B. The Benzene···Naphthalene Complex: A more Challenging System than the Benzene Dimer for newly Developed Computational Methods. J. Chem. Phys. 2015, 143, 114312. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Wang, Y.B. Highly Accurate Benchmark Calculations of the Interaction Energies in the Complexes C6H6···C6X6 (X = F, Cl, Br, and I). Int. J. Quantum Chem. 2017, 117, e25345. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Ng, C.-F.; Chow, H.-F.; Mak, T.C.W. Halogen-Bond-Mediated Assembly of a Single-Component Supramolecular Triangle and an Enantiomeric Pair of Double Helices from 2-(Iodoethynyl)pyridine Derivatives. Angew. Chem. Int. Ed. 2018, 57, 4986–4990. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-Z.; Koppireddi, S.; Wang, H.; Zhang, D.-W.; Li, Z.-T. Halogen Bonding Directed Supramolecular Quadruple and Double Helices from Hydrogen-Bonded Arylamide Foldamers. Angew. Chem. Int. Ed. 2019, 58, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Koppireddi, S.; Liu, C.-Z.; Wang, H.; Zhang, D.-W.; Li, Z.-T. Halogen and Hydrogen Bonding-Driven Self-Assembly of Supramolecular Macrocycles and Double Helices from Hydrogen-Bonded Arylamide Foldamers. CrystEngComm 2019, 21, 2626–2630. [Google Scholar] [CrossRef]
- Liu, C.-Z.; Koppireddi, S.; Wang, H.; Zhang, D.-W.; Li, Z.-T. Halogen Bonding-Driven Formation of Supramolecular Macrocycles and Double Helix. Chin. Chem. Lett. 2019, 30, 953–956. [Google Scholar] [CrossRef]
- Xu, Y.; Hao, A.; Xing, P. X···X Halogen Bond-Induced Supramolecular Helices. Angew. Chem. Int. Ed. 2022, 61, e202113786. [Google Scholar]
- Pitoňák, M.; Neogrády, P.; Řezáč, J.; Jurečka, P.; Urban, M.; Hobza, P. Benzene Dimer: High-Level Wave Function and Density Functional Theory Calculations. J. Chem. Theory Comput. 2008, 4, 1829–1834. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Gierschner, J.; Lüer, L.; Milián-Medina, B.; Oelkrug, D.; Egelhaaf, H.-J. Highly Emissive H-Aggregates or Aggregation-Induced Emission Quenching? The Photophysics of All-Trans para-Distyrylbenzene. J. Phys. Chem. Lett. 2013, 4, 2686–2697. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Wang, Y.-B. The π···π Stacking Interactions between Homogeneous Dimers of C6FxI(6-x) (x = 0, 1, 2, 3, 4, and 5): A Comparative Study with the Halogen Bond. J. Phys. Chem. A 2012, 116, 12486–12491. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z. Bifurcated Halogen Bond-Driven Supramolecular Double Helices from 1,2-Dihalotetrafluorobenzene and 2,2′-Bi(1,8-naphthyridine). Crystals 2022, 12, 937. https://doi.org/10.3390/cryst12070937
Wang Z. Bifurcated Halogen Bond-Driven Supramolecular Double Helices from 1,2-Dihalotetrafluorobenzene and 2,2′-Bi(1,8-naphthyridine). Crystals. 2022; 12(7):937. https://doi.org/10.3390/cryst12070937
Chicago/Turabian StyleWang, Ziyu. 2022. "Bifurcated Halogen Bond-Driven Supramolecular Double Helices from 1,2-Dihalotetrafluorobenzene and 2,2′-Bi(1,8-naphthyridine)" Crystals 12, no. 7: 937. https://doi.org/10.3390/cryst12070937
APA StyleWang, Z. (2022). Bifurcated Halogen Bond-Driven Supramolecular Double Helices from 1,2-Dihalotetrafluorobenzene and 2,2′-Bi(1,8-naphthyridine). Crystals, 12(7), 937. https://doi.org/10.3390/cryst12070937