
Microscopic Phase-Field Simulation of γ′ Precipitation in Ni-Based Binary Alloys Coupled with CALPHAD Method

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methodology
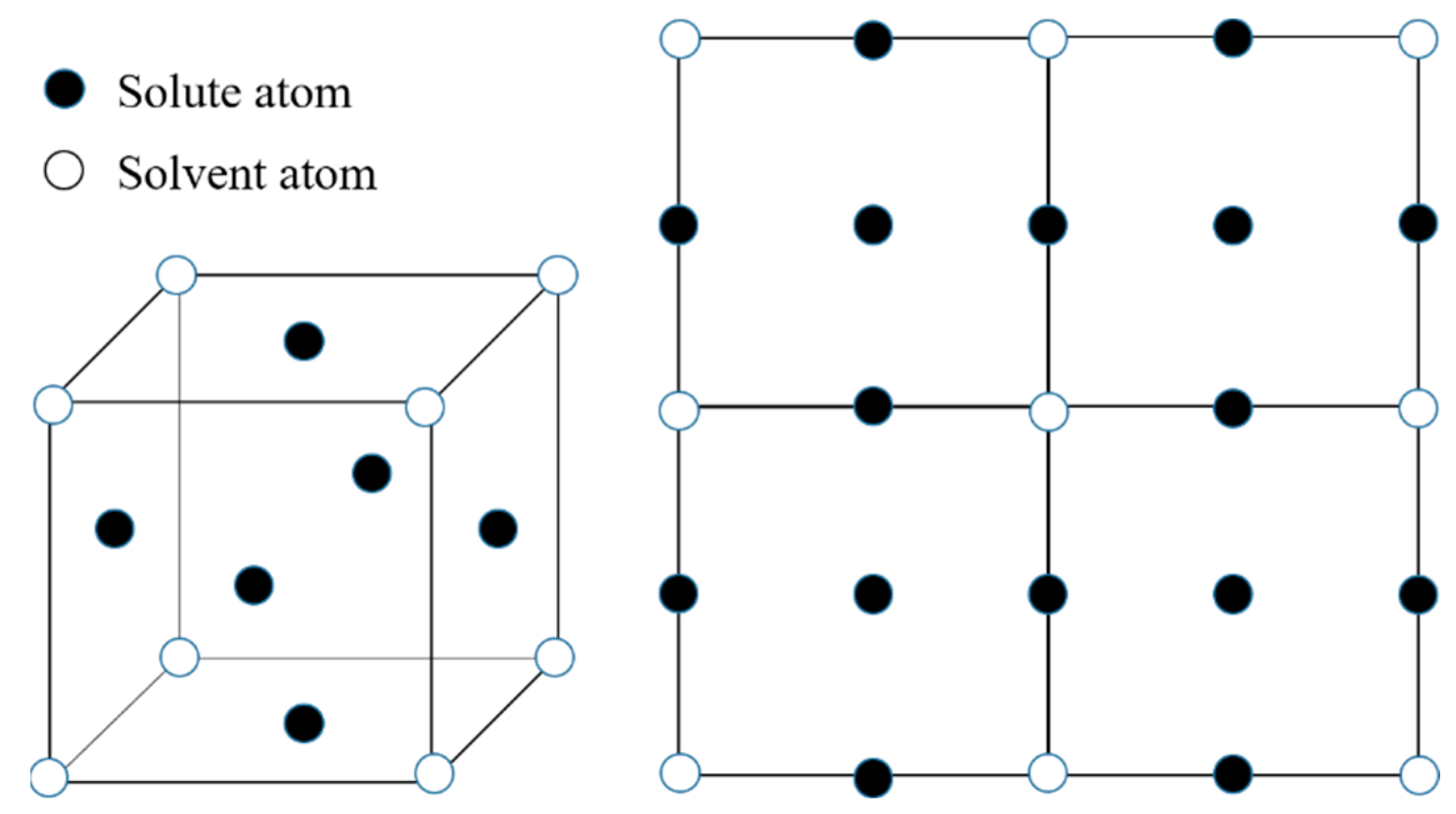
2.1. Concentration Wave Representation in γ Solid Solution
2.2. Helmholtz Free Energy
2.3. Order Parameter
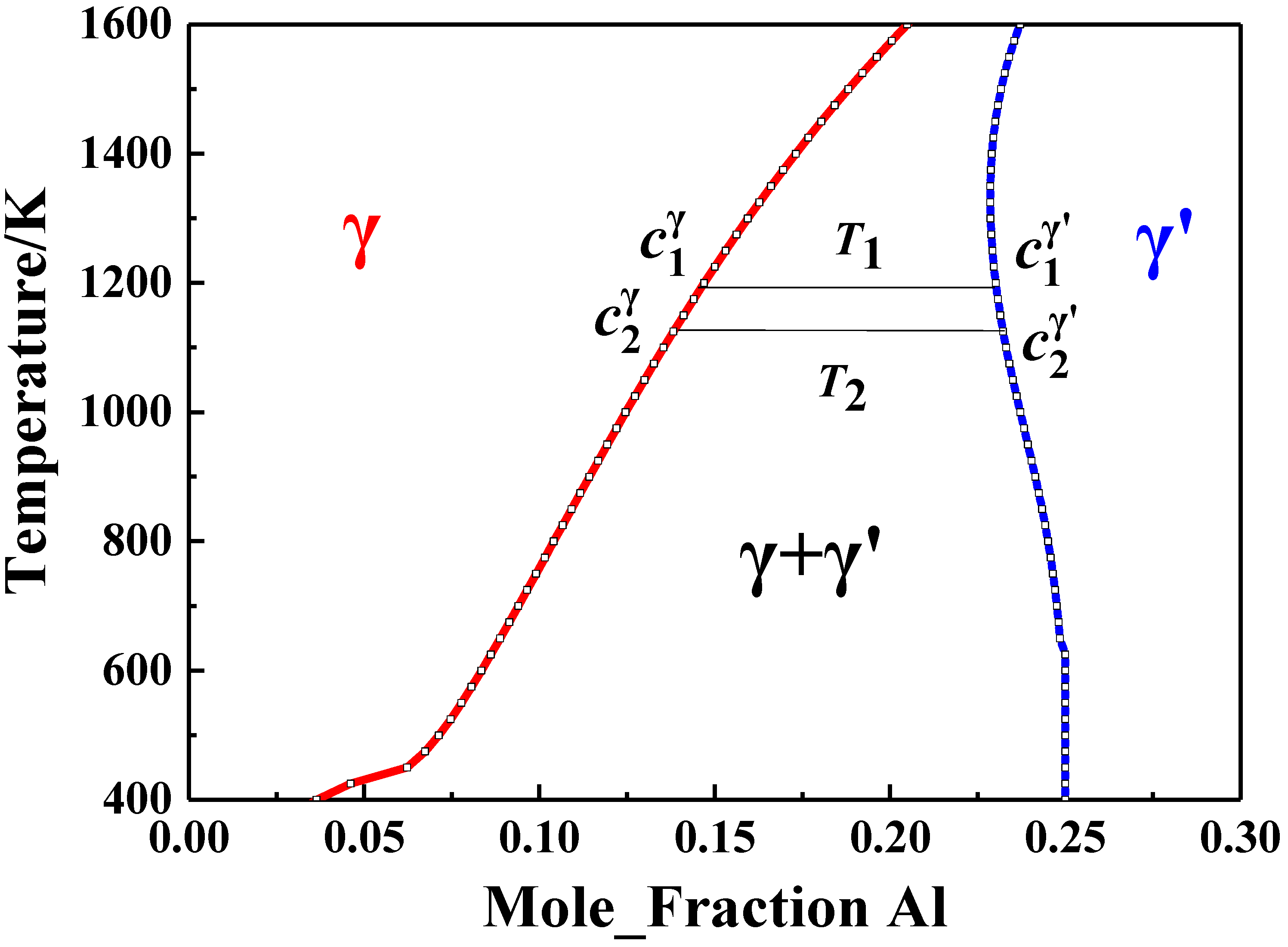
2.4. Coupling with CALPHAD to Calculate Interaction Energies
2.5. Kinetic Equations
3. Simulation Results and Discussion
3.1. 1st and 2nd Nearest-Neighbor Interaction Energies
3.2. Morphological Evolution of γ′ Precipitates
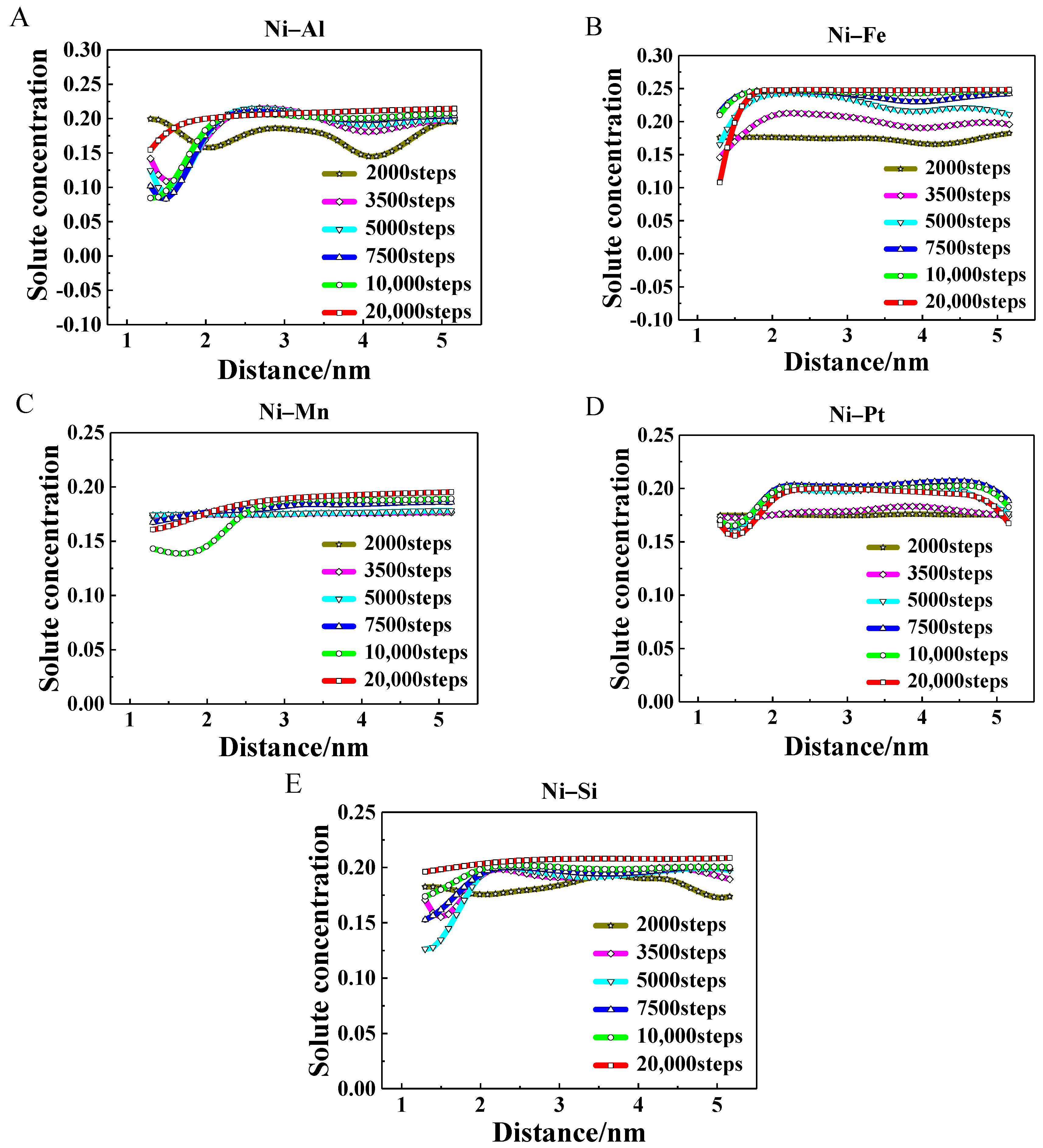
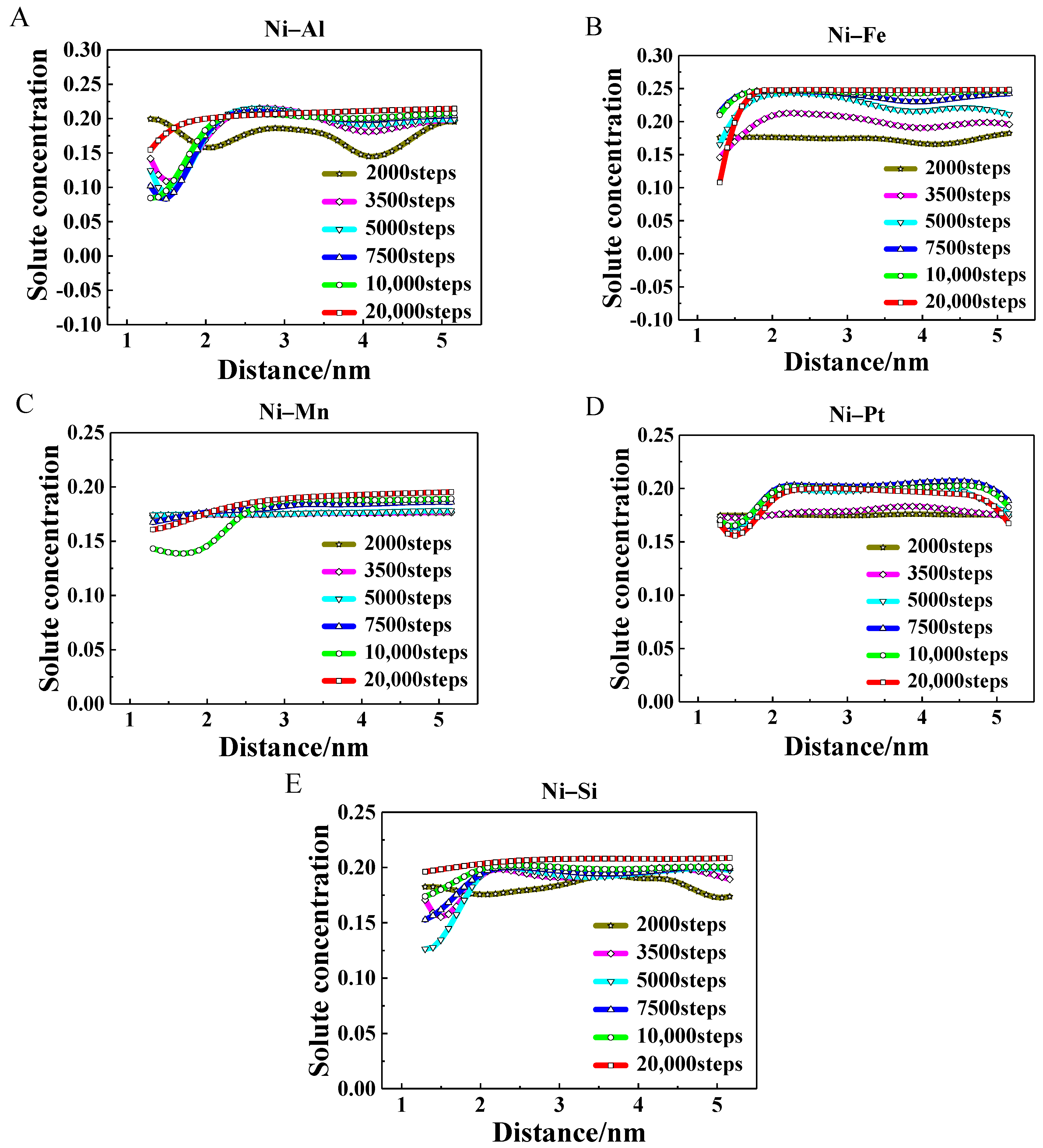
3.3. Evolution of Atomic Occupation
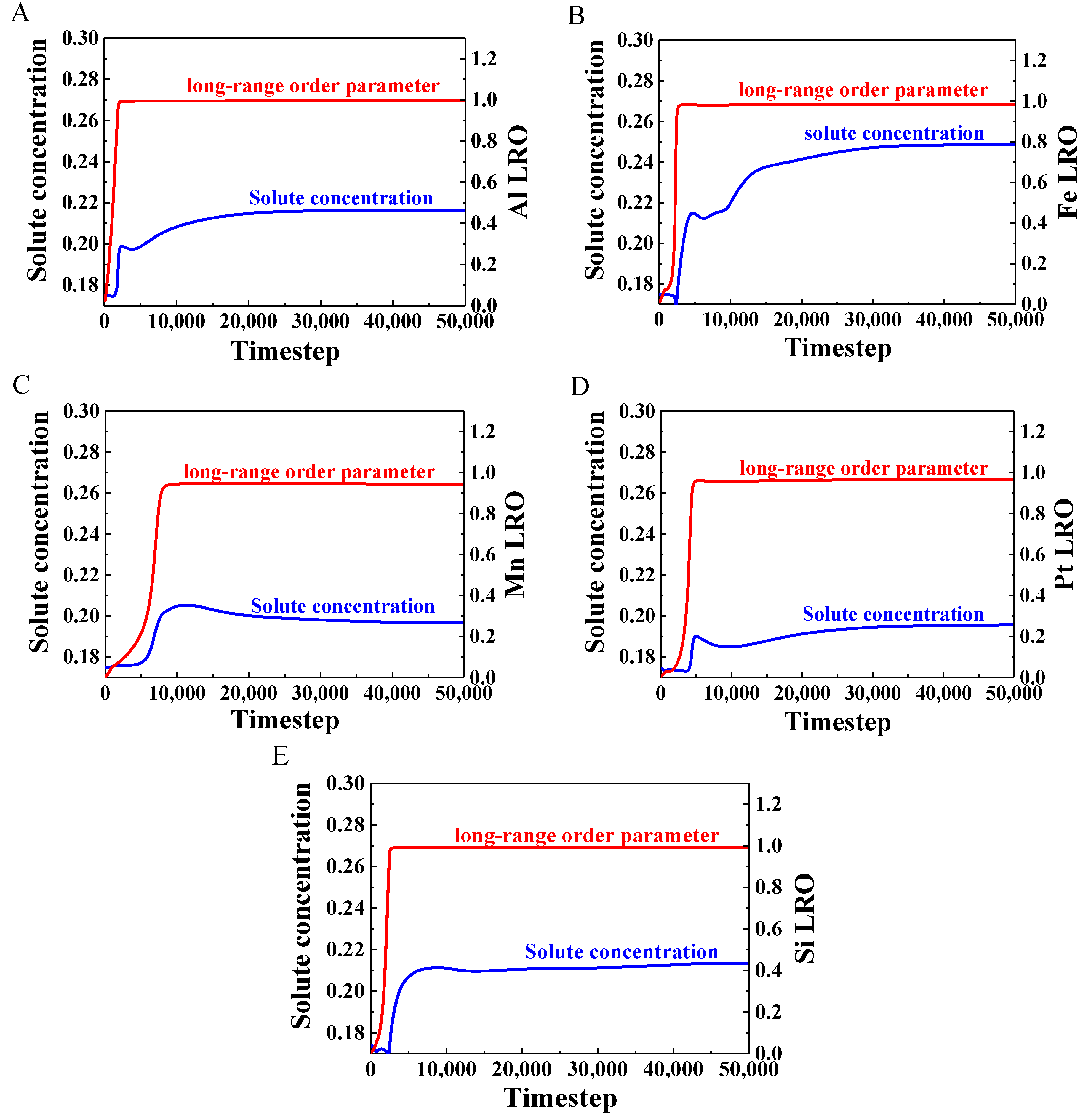
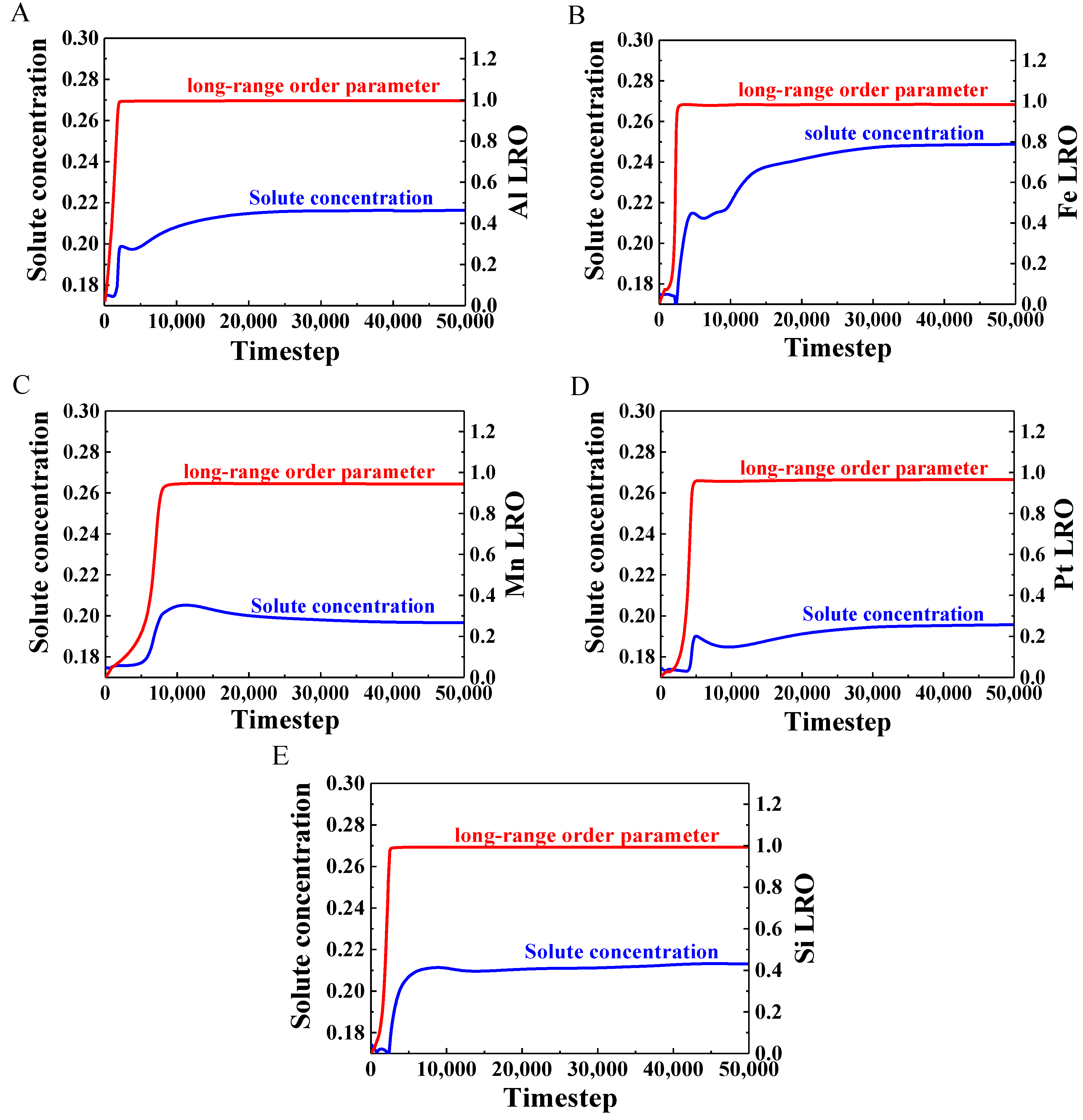
3.4. Evolution of Long-Range-Order Parameter and Solute Concentration
4. Conclusions
- There were different trends for the first and second nearest-neighbor interaction energies ( and ) in Ni–X (X = Al, Fe, Mn, Pt, or Si) alloys. The and values increased with temperature in the Ni–Al and Ni–Si alloys, while the opposite trend was seen in the Ni–Fe and Ni–Mn alloys. In the Ni–Pt alloy, the interaction energies first increased, with maxima at about 85 meV/atom for and −12 meV/atom for at 612 K, and then decreased with temperature.
- The morphological evolutions of the Ni–X alloys were simulated. The atomic ordering contributed to the γ′-phase precipitation from the γ phase, and the segregation and aggregation behavior of solute atoms in the γ phase greatly influenced the coarsening and growth process of the γ’ phase.
- The different occupation probabilities between solute and solvent atoms in the γ′ precipitates were predicted in the binary alloys. The solute atoms tend to occupy corner sites and the solvent atoms prefer to occupy the face sites. The equilibrium occupation probability values of Al, Mn, Pt, and Si atoms were close to 0.8, while the value for Fe atoms was close to 1.0.
- The temporal evolutions of the long-range parameter and the solute concentration were simulated to study the precipitation mechanism of the γ′ phase. The results showed that the long-range-order parameter (nearly 5000 timesteps) reached a steady state before the solute concentration (nearly 10,000 timesteps) in all alloys, and the clustering behavior revealed the migration of solute atoms during the precipitation process.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Soga, W.; Kaneno, Y.; Takasugi, T. Phase relation and microstructure in multi-phase intermetallic alloys based on Ni3Si–Ni3Ti–Ni3Nb pseudo-ternary alloy system. Intermetallics 2004, 12, 317–325. [Google Scholar]
- Wang, Y.; Banerjee, D.; Su, C.C.; Khachaturyan, A.G. Field kinetic model and computer simulation of precipitation of L12 ordered intermetallics from fcc solid solution. Acta Mater. 1998, 46, 2983–3001. [Google Scholar] [CrossRef]
- Sheu, H.H.; Hsiung, L.C.; Sheu, J.R. Synthesis of multiphase intermetallic compounds by mechanical alloying in Ni–Al–Ti system. J. Alloys Compd. 2009, 469, 483–487. [Google Scholar] [CrossRef]
- Savin, O.V.; Stepanova, N.N.; Akshentsev, Y.N.; Rodionov, D.P. Ordering kinetics in ternary Ni3Al–X alloys. Scr. Mater. 2001, 45, 883–888. [Google Scholar] [CrossRef]
- Chen, L.Q. A computer simulation technique for spinodal decomposition and ordering in ternary systems. Scr. Metall. Mater. 1993, 29, 683–688. [Google Scholar] [CrossRef]
- Bendersky, L.A.; Biancaniello, F.S.; Williams, M.E. Evolution of the two-phase microstructure L12 + DO22 in near-eutectoid Ni3(Al,V) alloy. J. Mater. Res. 1994, 9, 3068–3082. [Google Scholar] [CrossRef]
- Wu, R.; Zhao, Y.; Yin, Q.; Wang, J.; Wen, Z. Atomistic simulation studies of Ni-based superalloys. J. Alloys Compd. 2021, 855, 157355. [Google Scholar] [CrossRef]
- Wu, R.; Yin, Q.; Wang, J.; Mao, Q.; Wen, Z. Effect of Re on mechanical properties of single crystal Ni-based superalloys: Insights from first-principle and molecular dynamics. J. Alloys Compd. 2021, 862, 158643. [Google Scholar] [CrossRef]
- Wu, R.; Zhao, Y.; Liu, Y.; Ai, X. High temperature creep mechanisms of a single crystal superalloy: A phase-field simulation and microstructure characterization. Prog. Nat. Sci. 2020, 30, 366–370. [Google Scholar] [CrossRef]
- Cermak, J.; Rothova, V. Concentration dependence of ternary interdiffusion coefficients in Ni3Al/Ni3Al–X couples with X= Cr, Fe, Nb and Ti. Acta Mater. 2003, 51, 4411–4421. [Google Scholar] [CrossRef]
- Khachaturyan, A.G. Theory of structural transformations in solids. Mat. Res. Bull. 1984, 99, 125–127. [Google Scholar]
- Zhao, Y.; Zheng, C.; Li, X. Computer simulation of strain-induced morphological transformation of coherent precipitates. Int. J. Miner. Metall. Mater. 2003, 10, 55–60. [Google Scholar]
- Chen, L.Q. Computer simulation of structural transformations during precipitation of an ordered intermetallic phase. Acta Metall. Mater. 1991, 39, 2533–2551. [Google Scholar] [CrossRef]
- Li, X.L. Computer simulation for early precipitation of δ′(Al3Li). Prog. Nat. Sci. 2002, 12, 74. [Google Scholar]
- Poduri, R.; Chen, L.Q. Computer simulation of atomic ordering and compositional clustering in the pseudobinary Ni3Al–Ni3V system. Acta Mater. 1998, 46, 1719–1729. [Google Scholar] [CrossRef]
- Lu, X.G.; Wang, Z.; Cui, Y.; Jin, Z. Computational thermodynamics, computational kinetics, and materials design. Sci. Bull. 2014, 59, 1662–1671. [Google Scholar] [CrossRef]
- Mishin, Y.; Lozovoi, A.Y. Angular-dependent interatomic potential for tantalum. Acta Mater. 2006, 54, 5013–5026. [Google Scholar] [CrossRef]
- Kim, H.K.; Jung, W.S.; Lee, B.J. Modified embedded-atom method interatomic potentials for the Nb-C, Nb-N, Fe-Nb-C, and Fe-Nb-N system. J. Mater. Res. 2010, 25, 1288–1297. [Google Scholar] [CrossRef]
- Chen, L.Q.; Khachaturyan, A.G. Computer simulation of decomposition reactions accompanied by a congruent ordering of the second kind. Scr. Metall. Mater. 1991, 25, 61–66. [Google Scholar] [CrossRef]
- Khachaturyan, A.G. Nonlinear integral equations and their application to the problem of orderable alloys. Sov. Phys. Solid State. 1963, 5, 16–22. [Google Scholar]
- Khachaturyan, A.G. The problem of symmetry in statistical thermodynamics of substitutional and interstitial ordered solid solutions. Phys. Status Solidi-B 1973, 60, 9–37. [Google Scholar] [CrossRef]
- Andersson, J.O.; Helander, T.; Hglund, L. Thermo-Calc & DICTRA, computational tools for materials science. Calphad 2002, 26, 273–312. [Google Scholar]
- Wang, Y.; Chen, L.Q.; Khachaturyan, A.G. Kinetics of strain-induced morphological transformation in cubic alloys with a miscibility gap. Acta Metall. Mater. 1993, 41, 279–296. [Google Scholar] [CrossRef]
- Chen, L.Q.; Simmons, J.A. Microscopic master equation approach to diffusion transformations in inhomogeneous systems-single-site approximation and direct exchange mechanism. Acta Metall. Mater. 1994, 42, 2943–2954. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Zhao, Y.; Zhang, X.; Lu, X.-G.; Wang, C.; Zhang, Y. Microscopic Phase-Field Simulation of γ′ Precipitation in Ni-Based Binary Alloys Coupled with CALPHAD Method. Crystals 2022, 12, 971. https://doi.org/10.3390/cryst12070971
Liu Z, Zhao Y, Zhang X, Lu X-G, Wang C, Zhang Y. Microscopic Phase-Field Simulation of γ′ Precipitation in Ni-Based Binary Alloys Coupled with CALPHAD Method. Crystals. 2022; 12(7):971. https://doi.org/10.3390/cryst12070971
Chicago/Turabian StyleLiu, Zhenzhi, Yan Zhao, Xuyu Zhang, Xiao-Gang Lu, Chuanjun Wang, and Yu Zhang. 2022. "Microscopic Phase-Field Simulation of γ′ Precipitation in Ni-Based Binary Alloys Coupled with CALPHAD Method" Crystals 12, no. 7: 971. https://doi.org/10.3390/cryst12070971
APA StyleLiu, Z., Zhao, Y., Zhang, X., Lu, X.-G., Wang, C., & Zhang, Y. (2022). Microscopic Phase-Field Simulation of γ′ Precipitation in Ni-Based Binary Alloys Coupled with CALPHAD Method. Crystals, 12(7), 971. https://doi.org/10.3390/cryst12070971