
Simplified Approach to Characterize the Cooling Crystallization in a Modular Mini-Plant



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Substance System
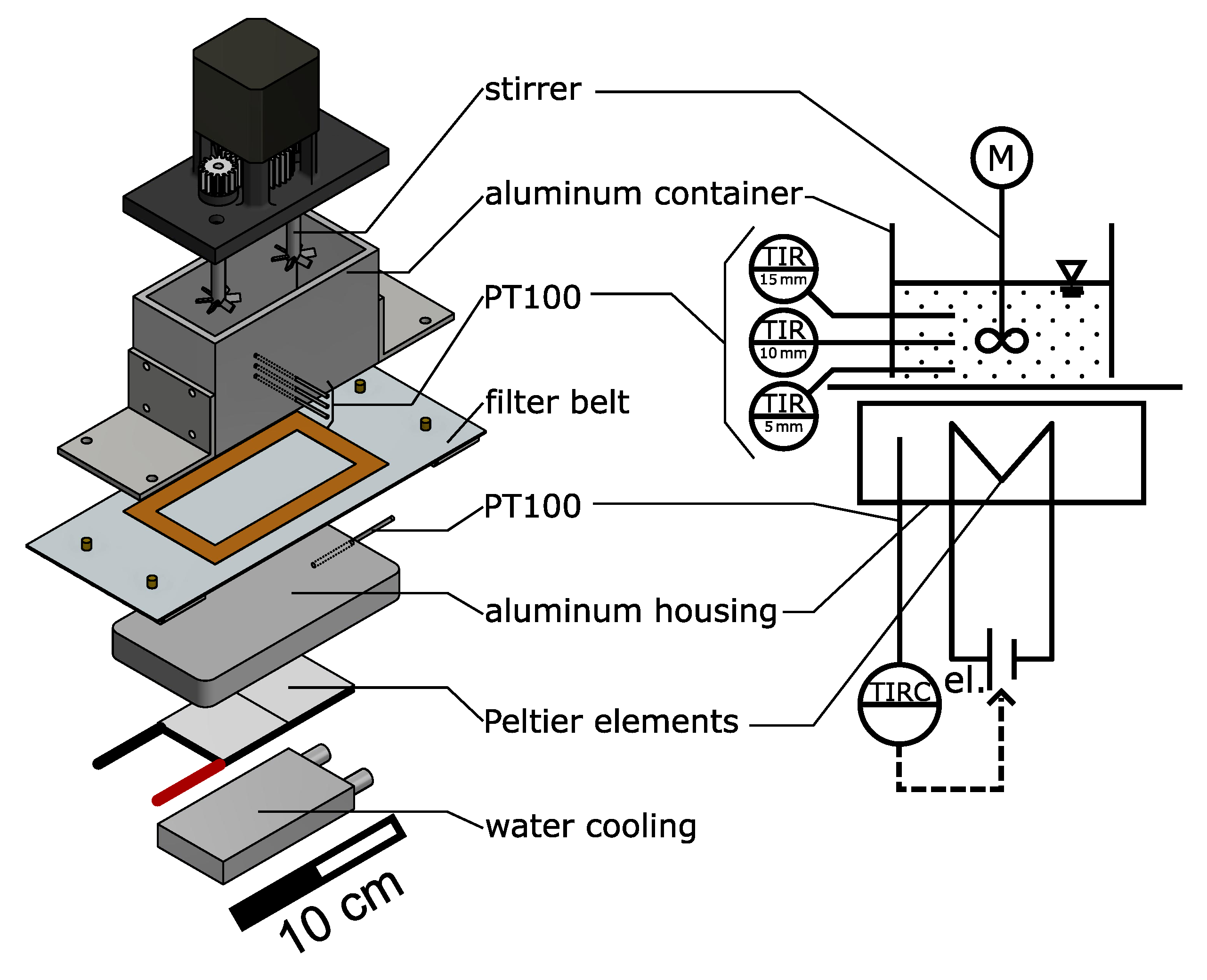
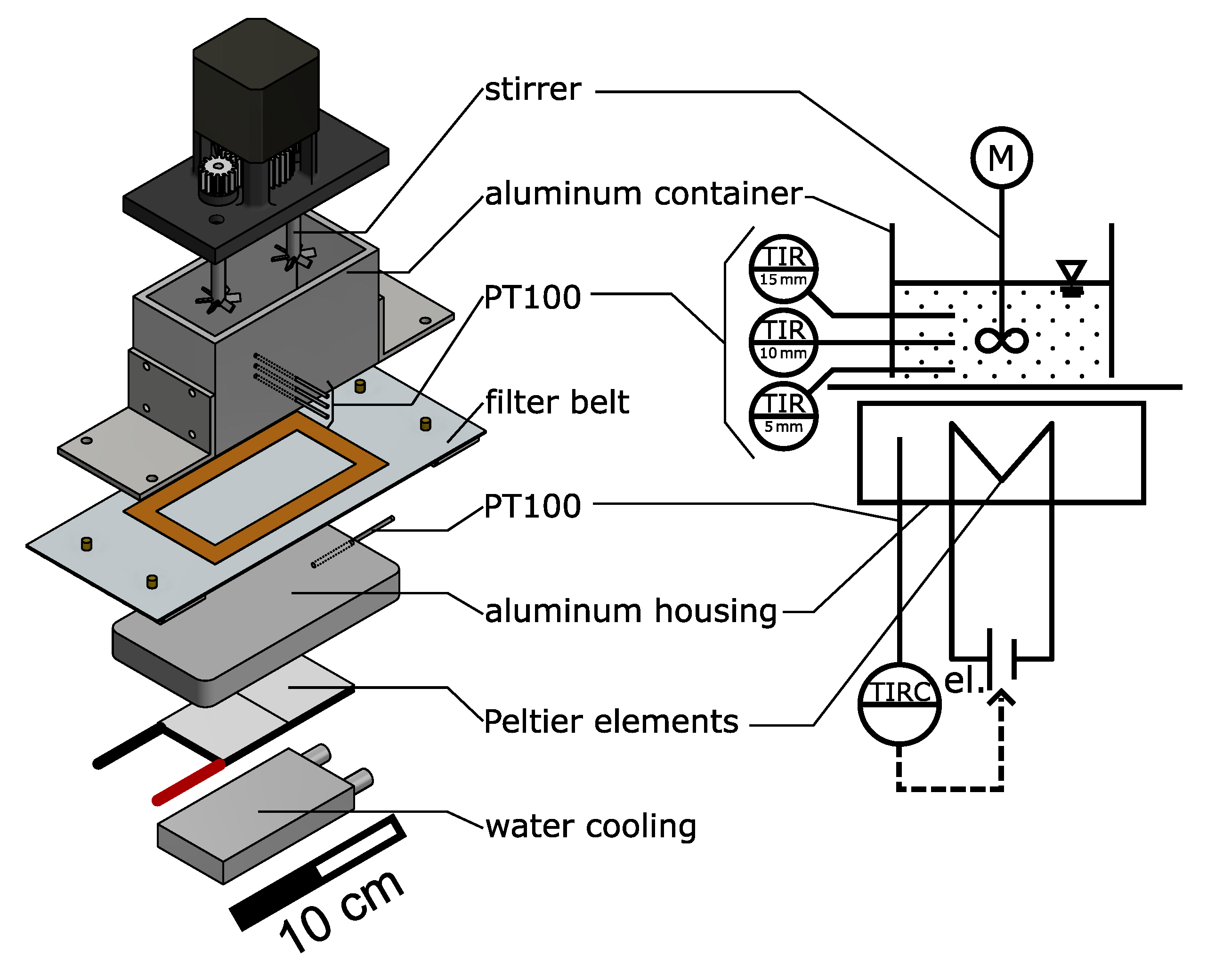
2.2. Process Container
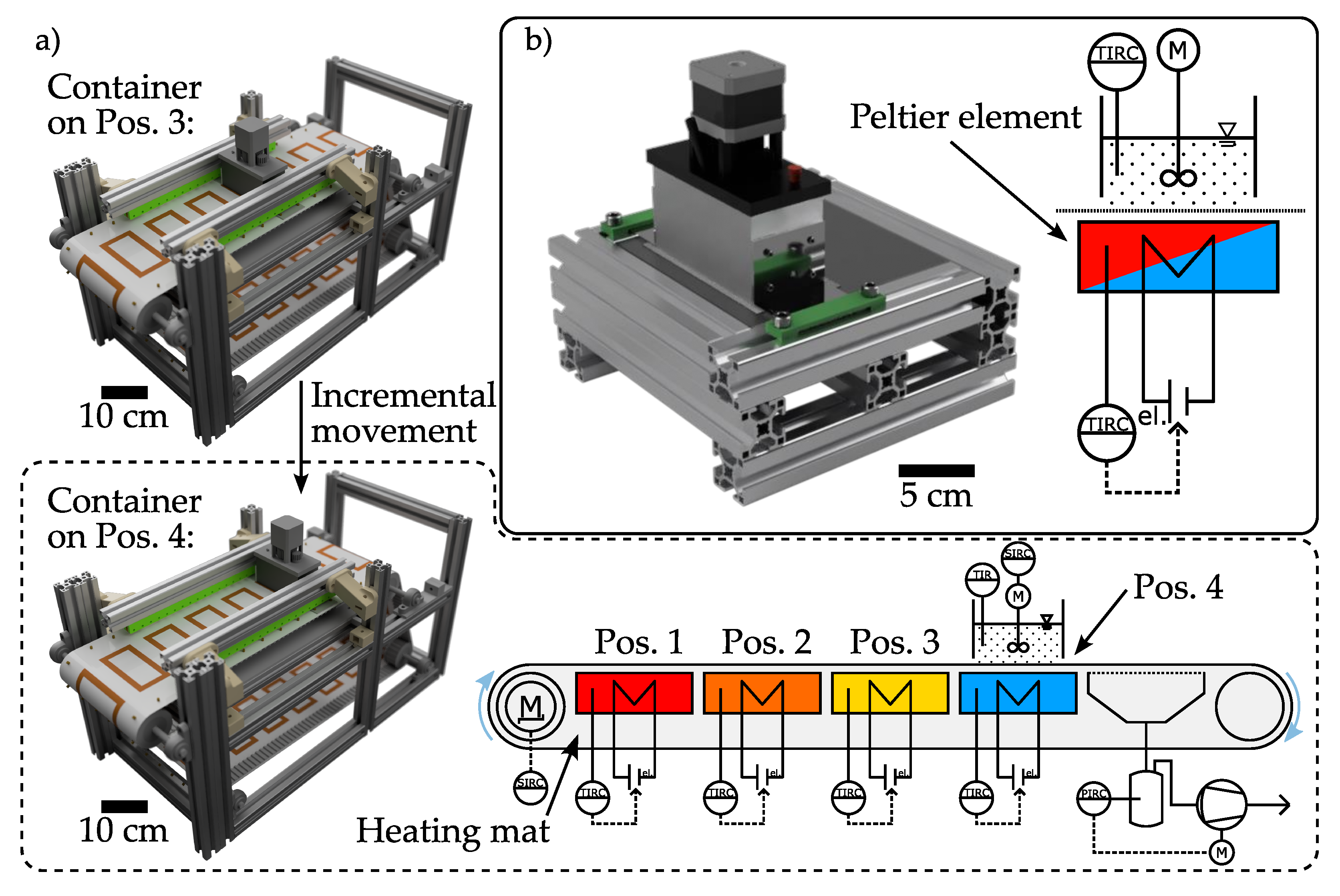
2.3. Quasi-Continuous Filter Belt Crystallizer: Mini-Plant
2.4. Simplified Benchtop Setup
2.5. Experimental Procedure
2.6. Analytics
3. Results and Discussion
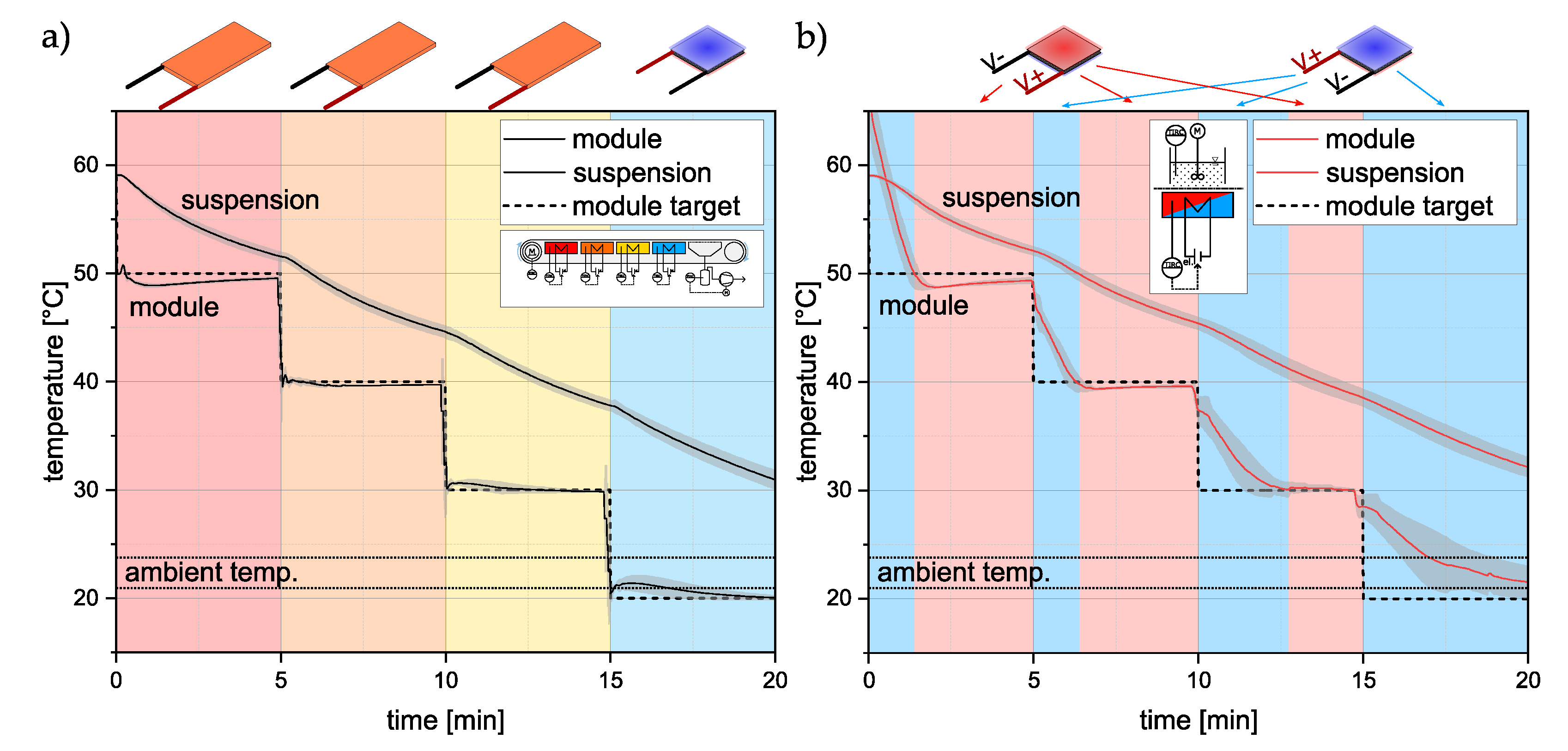
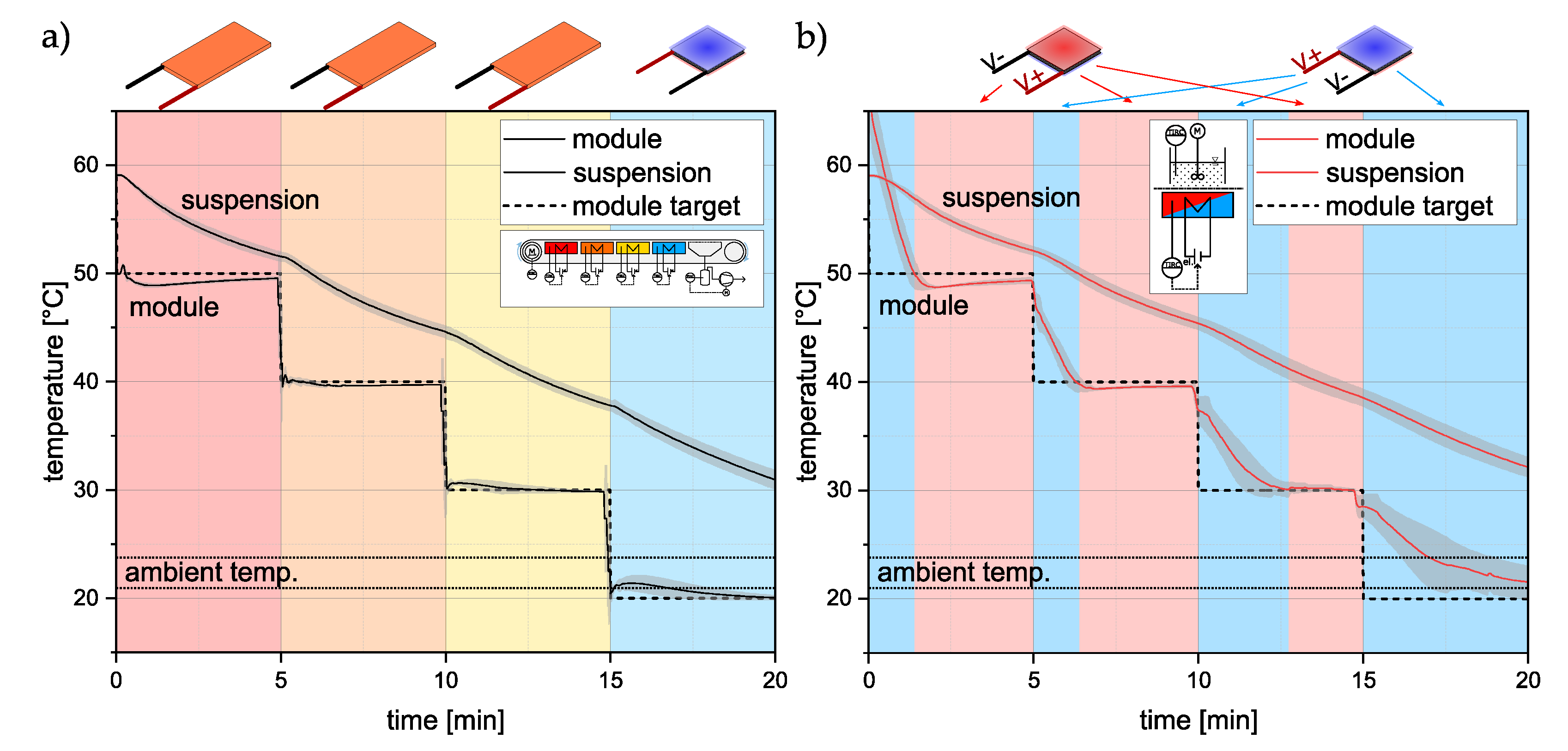
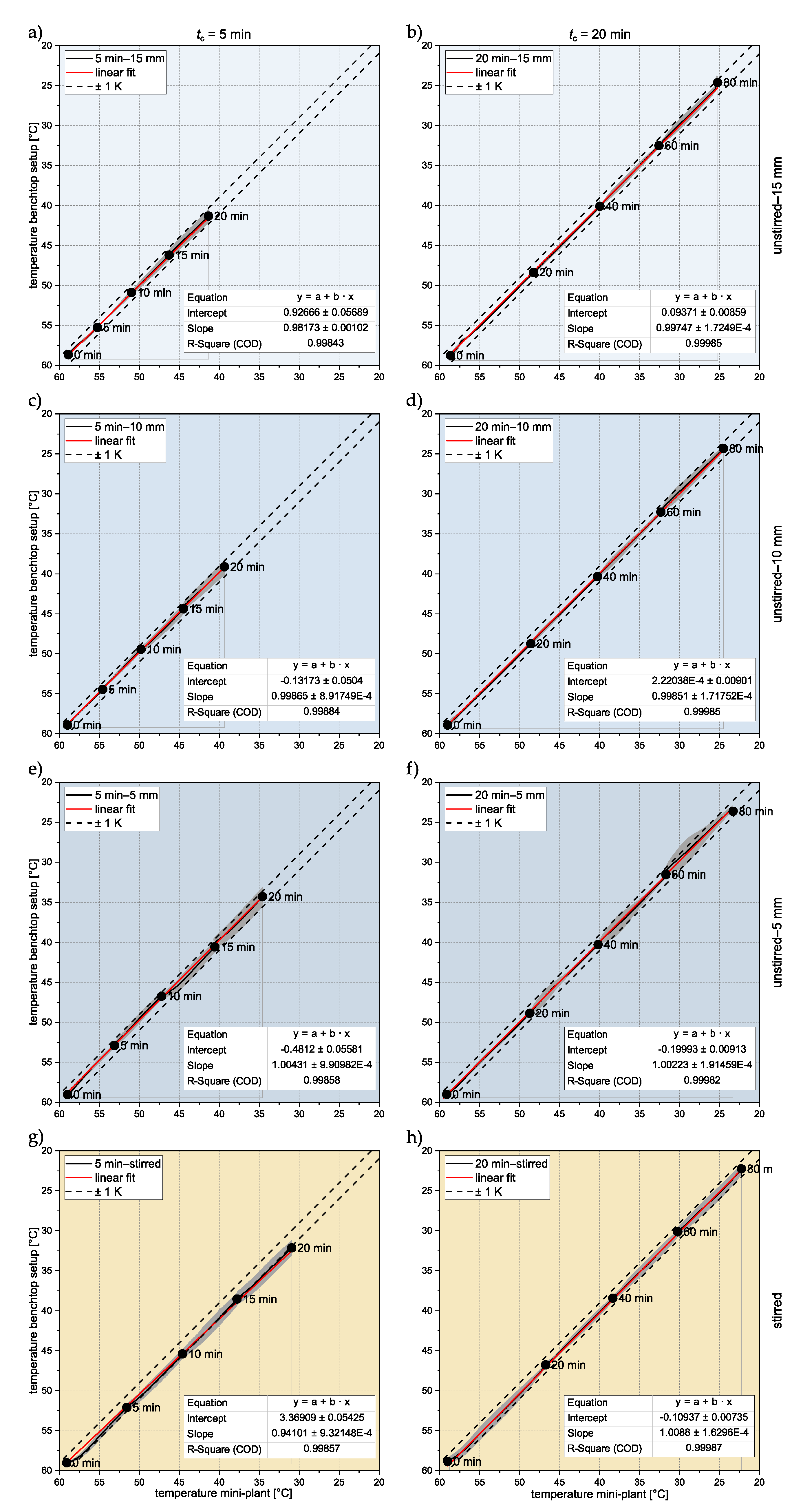
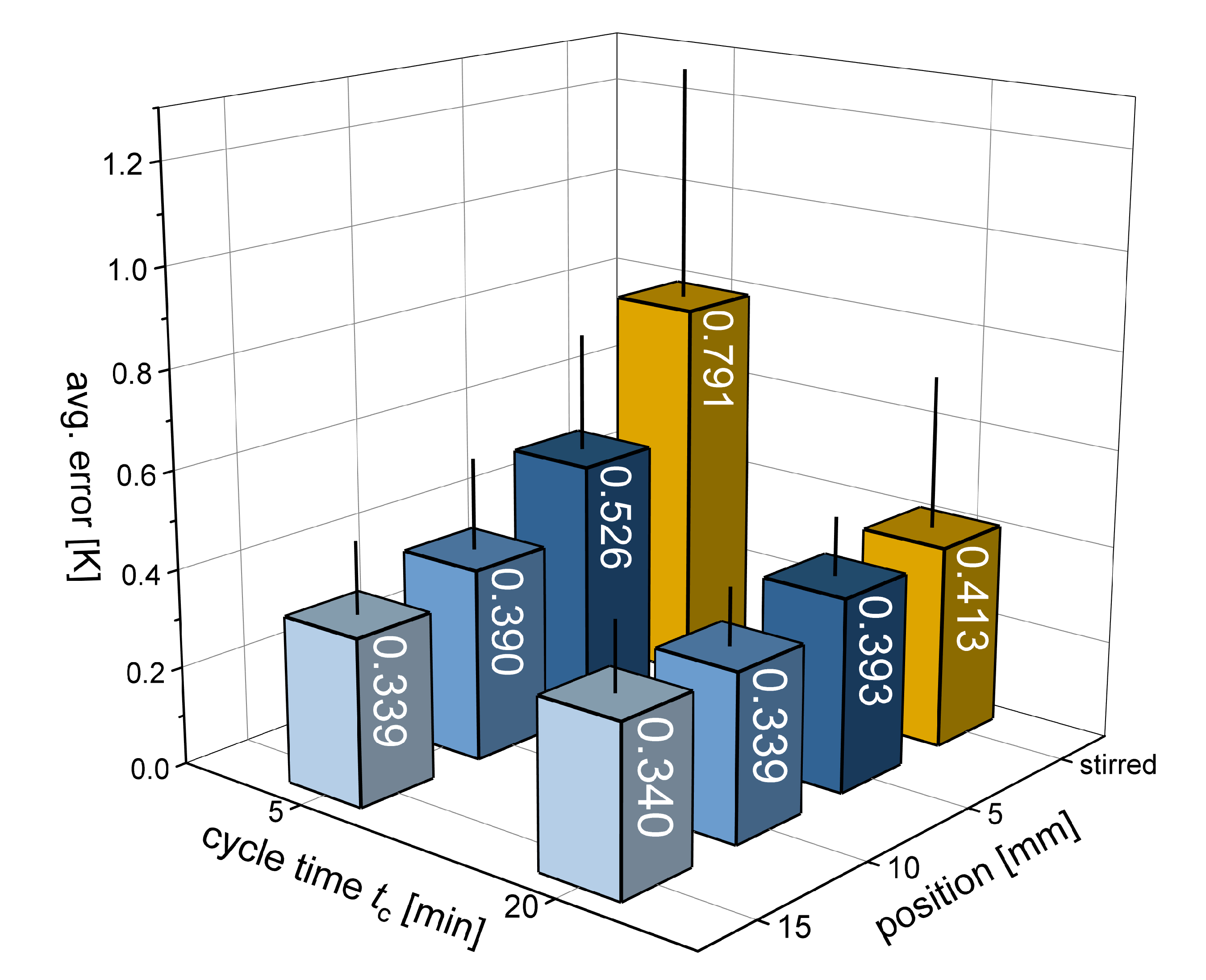
3.1. Temperature Profiles
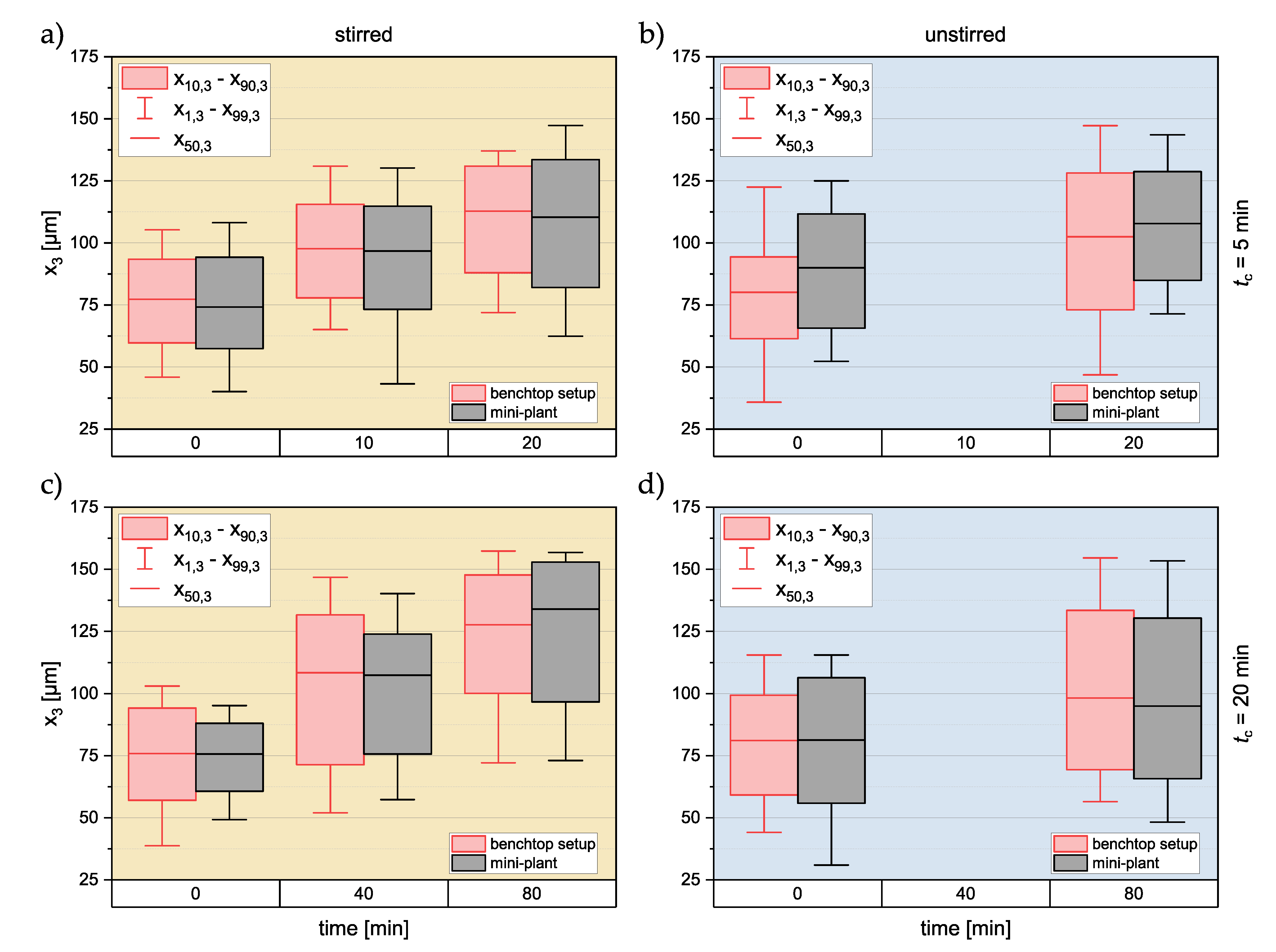
3.2. Product Properties
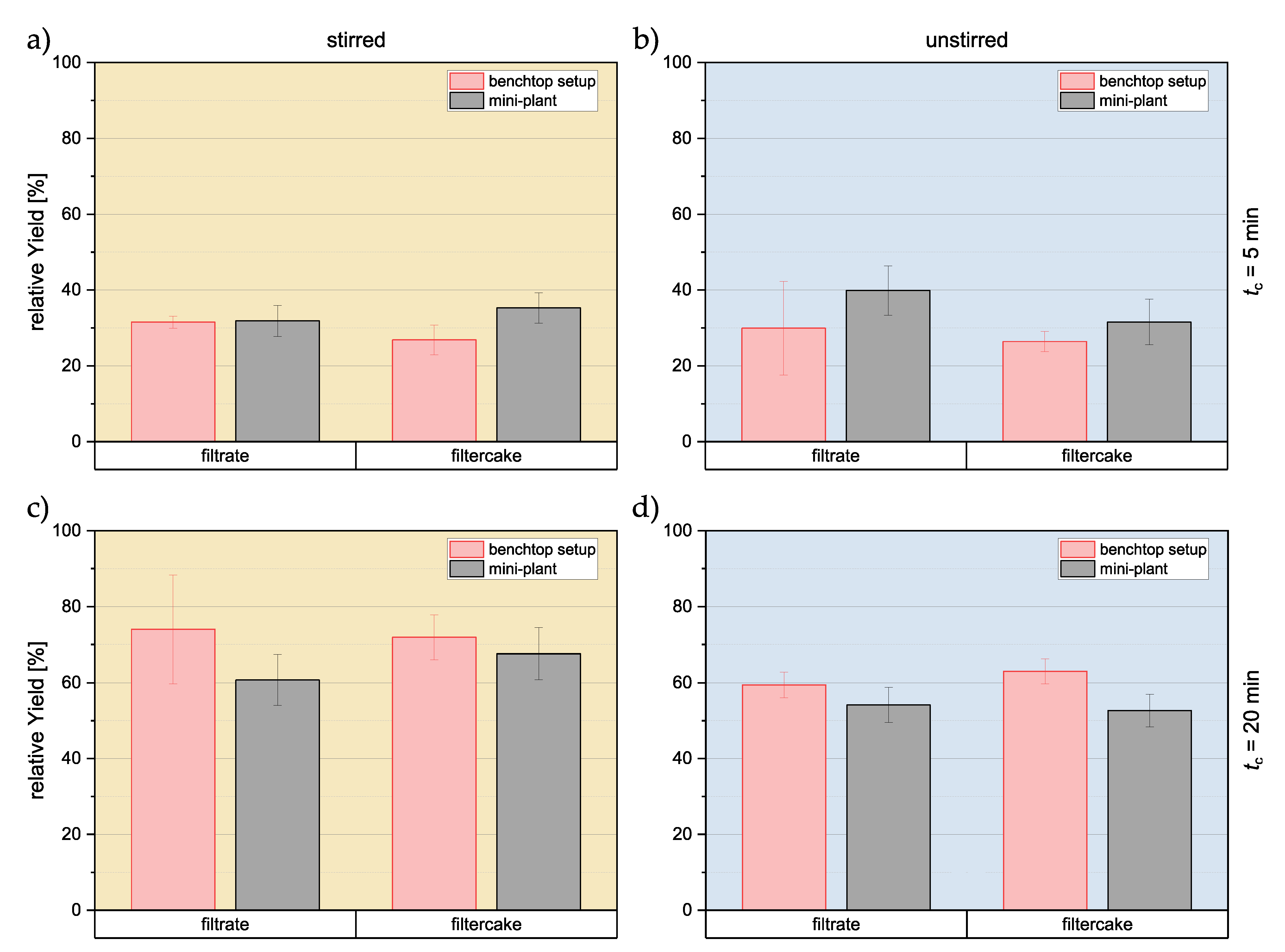
3.2.1. Relative Yield
3.2.2. Crystal Size Distribution
3.3. Quasi-Continuous Operation and Processing
4. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| API | Active Pharmaceutical Ingredient |
| CSD | Crystal Size Distribution |
| CVSF | Continuous Vacuum Screw Filter |
| Excess Crystal Mass | |
| EX | Explosion Protection |
| Dynamic Viscosity [kg·(m·s)] | |
| Diameter [m] | |
| g | Gravity Constant [m·s] |
| m | Mass [kg] |
| MSMPR | Mixed Suspension Mixed Product Removal |
| Density [kg·m] | |
| [kg·kg] | |
| QCFBC | Quasi Continuous Filter Belt Crystallizer |
| T | Temperatur [°C] |
| T | Saturation Temperature [°C] |
| t | Cycle Time [min] |
| Water Content [kg·kg ] | |
| crystal diameter [µm] | |
| Yield [%] |
References
- Mersmann, A. Crystallization Technology Handbook; Marcel Dekker Inc.: Hoboken, NJ, USA, 2001. [Google Scholar]
- Wong, S.Y.; Tatusko, A.P.; Trout, B.L.; Myerson, A.S. Development of Continuous Crystallization Processes Using a Single-Stage Mixed-Suspension, Mixed-Product Removal Crystallizer with Recycle. Cryst. Growth Des. 2012, 12, 5701–5707. [Google Scholar] [CrossRef]
- Cote, A.; Erdemir, D.; Girard, K.P.; Green, D.A.; Lovette, M.A.; Sirota, E.; Nere, N.K. Perspectives on the Current State, Challenges, and Opportunities in Pharmaceutical Crystallization Process Development. Cryst. Growth Des. 2020, 20, 7568–7581. [Google Scholar] [CrossRef]
- Orehek, J.; Teslić, D.; Likozar, B. Continuous Crystallization Processes in Pharmaceutical Manufacturing: A Review. Org. Process. Res. Dev. 2021, 25, 16–42. [Google Scholar] [CrossRef]
- Testa, C.J.; Shvedova, K.; Hu, C.; Wu, W.; Born, S.C.; Takizawa, B.; Mascia, S. Heterogeneous Crystallization as a Process Intensification Technology in an Integrated Continuous Manufacturing Process for Pharmaceuticals. Org. Process. Res. Dev. 2021, 25, 225–238. [Google Scholar] [CrossRef]
- Wood, B.; Girard, K.P.; Polster, C.S.; Croker, D.M. Progress to Date in the Design and Operation of Continuous Crystallization Processes for Pharmaceutical Applications. Org. Process. Res. Dev. 2019, 23, 122–144. [Google Scholar] [CrossRef]
- Zhang, D.; Xu, S.; Du, S.; Wang, J.; Gong, J. Progress of Pharmaceutical Continuous Crystallization. Engineering 2017, 3, 354–364. [Google Scholar] [CrossRef]
- Domokos, A.; Nagy, B.; Szilágyi, B.; Marosi, G.; Nagy, Z.K. Integrated Continuous Pharmaceutical Technologies—A Review. Org. Process. Res. Dev. 2021, 25, 721–739. [Google Scholar] [CrossRef]
- Eren, A.; Civati, F.; Ma, W.; Gamekkanda, J.C.; Myerson, A.S. Continuous crystallization and its potential use in drug substance Manufacture: A review. J. Cryst. Growth 2023, 601, 126958. [Google Scholar] [CrossRef]
- Chen, J.; Sarma, B.; Evans, J.M.B.; Myerson, A.S. Pharmaceutical Crystallization. Cryst. Growth Des. 2011, 11, 887–895. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, G. Kristallisation in der Industriellen Praxis, 1st ed.; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Lee, S.L.; O’Connor, T.F.; Yang, X.; Cruz, C.N.; Chatterjee, S.; Madurawe, R.D.; Moore, C.M.V.; Yu, L.X.; Woodcock, J. Modernizing Pharmaceutical Manufacturing: From Batch to Continuous Production. J. Pharm. Innov. 2015, 10, 191–199. [Google Scholar] [CrossRef]
- Myerson, A.S. Handbook of Industrial Crystallization, 2nd ed.; Butterworth-Heinemann: Boston, MA, USA, 2002. [Google Scholar]
- Schmalenberg, M.; Kreis, S.; Weick, L.K.; Haas, C.; Sallamon, F.; Kockmann, N. Continuous Cooling Crystallization in a Coiled Flow Inverter Crystallizer Technology—Design, Characterization, and Hurdles. Processes 2021, 9, 1537. [Google Scholar] [CrossRef]
- Hohmann, L.; Löbnitz, L.; Menke, C.; Santhirakumaran, B.; Stier, P.; Stenger, F.; Dufour, F.; Wiese, G.; zur Horst-Meyer, S.; Kusserow, B.; et al. Continuous Downstream Processing of Amino Acids in a Modular Miniplant. Chem. Eng. Technol. 2018, 41, 1152–1164. [Google Scholar] [CrossRef]
- Besenhard, M.O.; Neugebauer, P.; Scheibelhofer, O.; Khinast, J.G. Crystal Engineering in Continuous Plug-Flow Crystallizers. Cryst. Growth Des. 2017, 17, 6432–6444. [Google Scholar] [CrossRef] [PubMed]
- Lührmann, M.C.; Timmermann, J.; Schembecker, G.; Wohlgemuth, K. Enhanced Product Quality Control through Separation of Crystallization Phenomena in a Four-Stage MSMPR Cascade. Cryst. Growth Des. 2018, 18, 7323–7334. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Kim, J.M.; Chang, S.M.; Kim, W.S. Taylor Vortex Effect on Phase Transformation of Guanosine 5-Monophosphate in Drowning-Out Crystallization. Ind. Eng. Chem. Res. 2010, 49, 4865–4872. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, W.S. Synthesis of Core–Shell Particles of Nickel–Manganese–Cobalt Hydroxides in a Continuous Couette-Taylor Crystallizer. Cryst. Growth Des. 2017, 17, 3677–3686. [Google Scholar] [CrossRef]
- Briggs, N.E.B.; Schacht, U.; Raval, V.; McGlone, T.; Sefcik, J.; Florence, A.J. Seeded Crystallization of β-l-Glutamic Acid in a Continuous Oscillatory Baffled Crystallizer. Org. Process. Res. Dev. 2015, 19, 1903–1911. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Lu, H.; Wang, J.; Xiao, Y.; Zhou, Y.; Bao, Y.; Hao, H. Recent progress of continuous crystallization. J. Ind. Eng. Chem. 2017, 54, 14–29. [Google Scholar] [CrossRef]
- Steenweg, C.; Kufner, A.C.; Habicht, J.; Wohlgemuth, K. Towards Continuous Primary Manufacturing Processes—Particle Design through Combined Crystallization and Particle Isolation. Processes 2021, 9, 2187. [Google Scholar] [CrossRef]
- Acevedo, D.; Peña, R.; Yang, Y.; Barton, A.; Firth, P.; Nagy, Z.K. Evaluation of mixed suspension mixed product removal crystallization processes coupled with a continuous filtration system. Chem. Eng. Process. Process. Intensif. 2016, 108, 212–219. [Google Scholar] [CrossRef]
- Liu, Y.C.; Domokos, A.; Coleman, S.; Firth, P.; Nagy, Z.K. Development of Continuous Filtration in a Novel Continuous Filtration Carousel Integrated with Continuous Crystallization. Org. Process. Res. Dev. 2019, 23, 2655–2665. [Google Scholar] [CrossRef]
- Ottoboni, S.; Price, C.J.; Steven, C.; Meehan, E.; Barton, A.; Firth, P.; Mitchell, A.; Tahir, F. Development of a Novel Continuous Filtration Unit for Pharmaceutical Process Development and Manufacturing. J. Pharm. Sci. 2019, 108, 372–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domokos, A.; Nagy, B.; Gyürkés, M.; Farkas, A.; Tacsi, K.; Pataki, H.; Liu, Y.C.; Balogh, A.; Firth, P.; Szilágyi, B.; et al. End-to-end continuous manufacturing of conventional compressed tablets: From flow synthesis to tableting through integrated crystallization and filtration. Int. J. Pharm. 2020, 581, 119297. [Google Scholar] [CrossRef] [PubMed]
- Nagy, B.; Szilágyi, B.; Domokos, A.; Tacsi, K.; Pataki, H.; Marosi, G.; Nagy, Z.K.; Nagy, Z.K. Modeling of pharmaceutical filtration and continuous integrated crystallization-filtration processes. Chem. Eng. J. 2021, 413, 127566. [Google Scholar] [CrossRef]
- Mascia, S.; Heider, P.L.; Zhang, H.; Lakerveld, R.; Benyahia, B.; Barton, P.I.; Braatz, R.D.; Cooney, C.L.; Evans, J.M.B.; Jamison, T.F.; et al. End-to-End Continuous Manufacturing of Pharmaceuticals: Integrated Synthesis, Purification, and Final Dosage Formation. Angew. Chem. Int. Ed. 2013, 52, 12359–12363. [Google Scholar] [CrossRef]
- Hu, C.; Testa, C.J.; Born, S.C.; Wu, W.; Shvedova, K.; Sayin, R.; Halkude, B.S.; Casati, F.; Ramnath, A.; Hermant, P.; et al. E-factor analysis of a pilot plant for end-to-end integrated continuous manufacturing (ICM) of pharmaceuticals. Green Chem. 2020, 22, 4350–4356. [Google Scholar] [CrossRef]
- Steenweg, C.; Seifert, A.I.; Schembecker, G.; Wohlgemuth, K. Characterization of a Modular Continuous Vacuum Screw Filter for Small-Scale Solid–Liquid Separation of Suspensions. Org. Process. Res. Dev. 2021, 25, 926–940. [Google Scholar] [CrossRef]
- Baldea, M.; Edgar, T.F.; Stanley, B.L.; Kiss, A.A. Modular manufacturing processes: Status, challenges, and opportunities. AIChE J. 2017, 63, 4262–4272. [Google Scholar] [CrossRef] [Green Version]
- Lier, S.; Paul, S.; Ferdinand, D.; Grünewald, M. Modulare Verfahrenstechnik: Apparateentwicklung für wandlungsfähige Produktionssysteme. Chem. Ing. Tech. 2016, 88, 1444–1454. [Google Scholar] [CrossRef]
- Dobler, T.; Buchheiser, S.; Gleiß, M.; Nirschl, H. Development and Commissioning of a Small-Scale, Modular and Integrated Plant for the Quasi-Continuous Production of Crystalline Particles. Processes 2021, 9, 663. [Google Scholar] [CrossRef]
- Höving, S.; Oldach, B.; Kockmann, N. Cooling Crystallization with Complex Temperature Profiles on a Quasi-Continuous and Modular Plant. Processes 2022, 10, 1047. [Google Scholar] [CrossRef]
- Wilson, A.J.C. Atlas der Zuckerkristalle—Atlas of Sugar Crystals by G. Vavrinecz. Acta Crystallogr. 1966, 20, 152. [Google Scholar] [CrossRef] [Green Version]
- Löbnitz, L. Auslegung des Separationsprozesses und Entwicklung neuer Verfahrenskonzepte zur Integrierten Produktion und Separation Kristalliner Aminosäuren; Karlsruher Institut für Technologie: Karlsruhe, Germany, 2020. [Google Scholar]
- Sonnenschein, J.; Hermes, M.; Höving, S.; Kockmann, N.; Wohlgemuth, K. Population balance modeling of unstirred cooling crystallization on an integrated belt filter. Comput. Chem. Eng. 2022, 167, 108024. [Google Scholar] [CrossRef]
- Pot, A. Industrial Sucrose Crystallisation: A Study on the Development of Crystal Size Distributions in Continuous and Batch Sucrose Suspension Crystallisers. Ph.D. Thesis, Delft University of Technology, Delft, The Netherlands, 1983. [Google Scholar]
- Deutsches Institut für Normung. Agitators and Baffles for Agitator Vessels; Types, Terms and Main Dimensions; Deutsches Institut für Normung: Berlin, Germany, 1992. [Google Scholar]
- Hohmann, L.; Greinert, T.; Mierka, O.; Turek, S.; Schembecker, G.; Bayraktar, E.; Wohlgemuth, K.; Kockmann, N. Analysis of Crystal Size Dispersion Effects in a Continuous Coiled Tubular Crystallizer: Experiments and Modeling. Cryst. Growth Des. 2018, 18, 1459–1473. [Google Scholar] [CrossRef]
- Schneider, F.; Schliephake, D.; Klimmek, A. Über die Viskosität von reinen Saccharoselösungen. Zucker 1963, 16, 465–473. [Google Scholar]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Höving, S.; Bolien, P.; Siebers, P.; Kockmann, N. Simplified Approach to Characterize the Cooling Crystallization in a Modular Mini-Plant. Crystals 2023, 13, 147. https://doi.org/10.3390/cryst13010147
Höving S, Bolien P, Siebers P, Kockmann N. Simplified Approach to Characterize the Cooling Crystallization in a Modular Mini-Plant. Crystals. 2023; 13(1):147. https://doi.org/10.3390/cryst13010147
Chicago/Turabian StyleHöving, Stefan, Phil Bolien, Paul Siebers, and Norbert Kockmann. 2023. "Simplified Approach to Characterize the Cooling Crystallization in a Modular Mini-Plant" Crystals 13, no. 1: 147. https://doi.org/10.3390/cryst13010147
APA StyleHöving, S., Bolien, P., Siebers, P., & Kockmann, N. (2023). Simplified Approach to Characterize the Cooling Crystallization in a Modular Mini-Plant. Crystals, 13(1), 147. https://doi.org/10.3390/cryst13010147