
Structural Determination of the Hexacoordinated [Zn(L)2]2+ Complex Isomer Type Using Solution-State NMR, DFT Calculations and X-ray Crystallography



Abstract
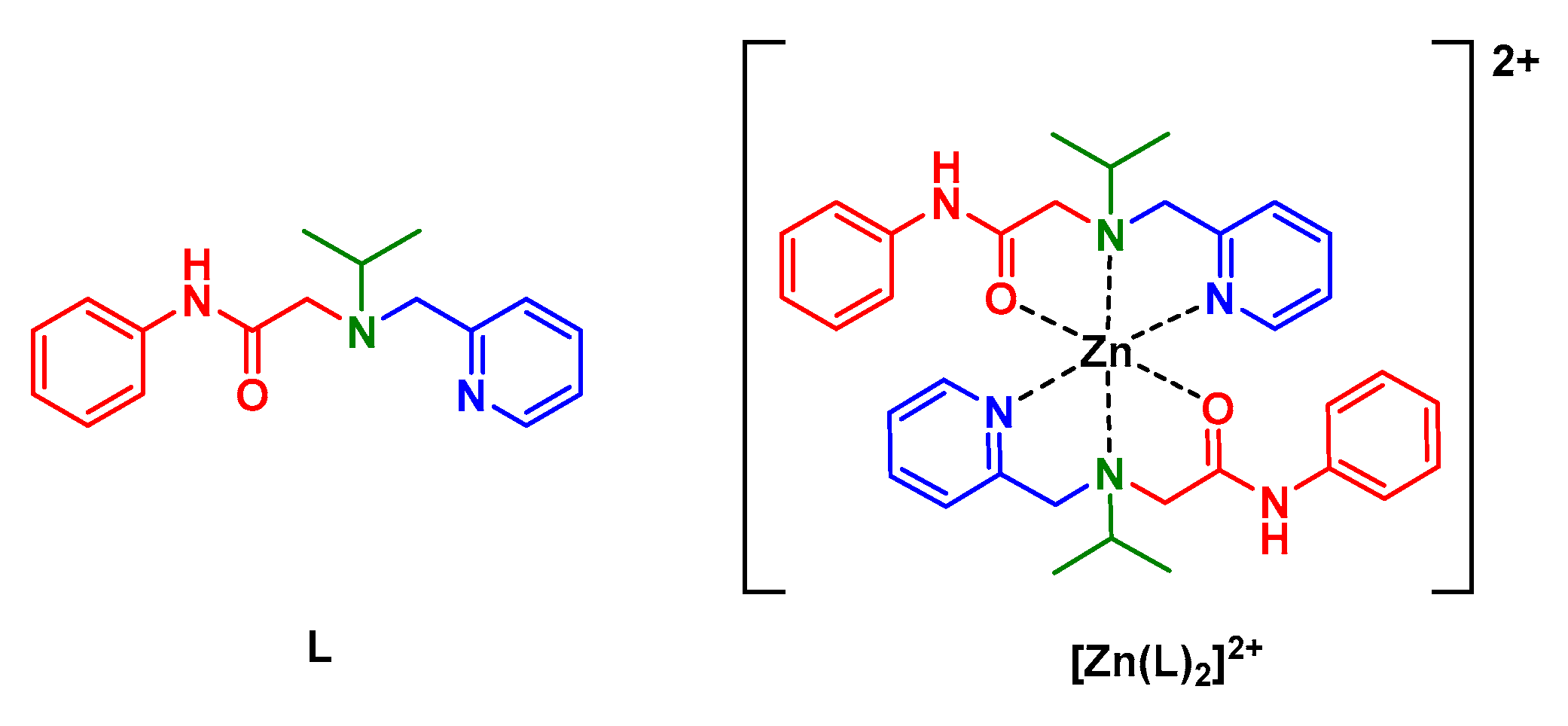
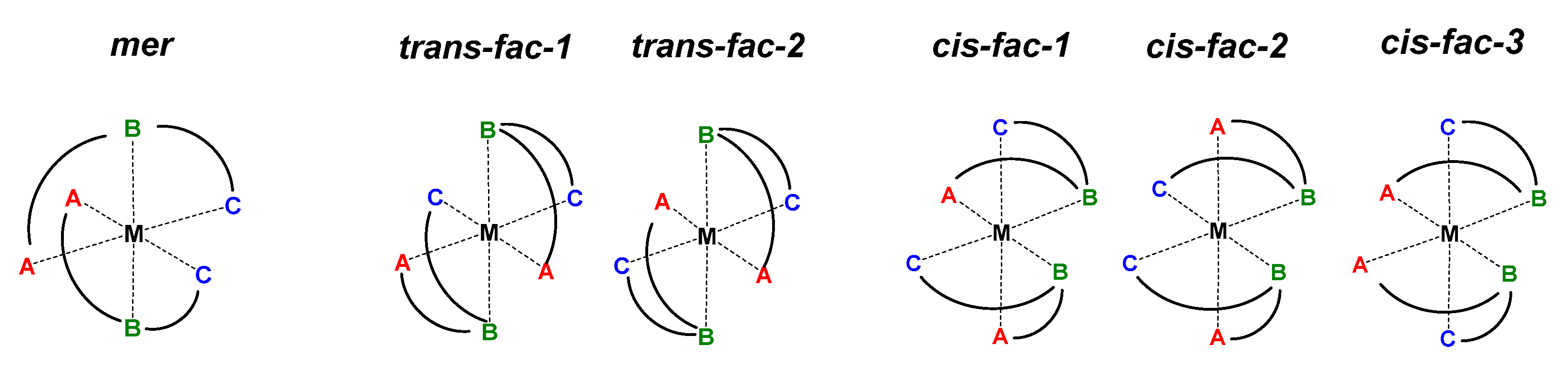
:1. Introduction
K = Σh|Fhc|/Σh|Fho|
2. Materials and Methods
2.1. Synthesis and Characterisation
2.1.1. General Remarks
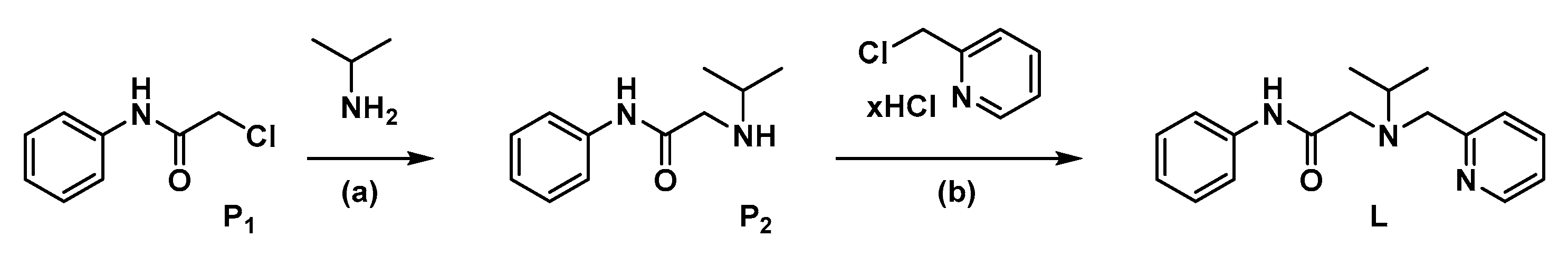
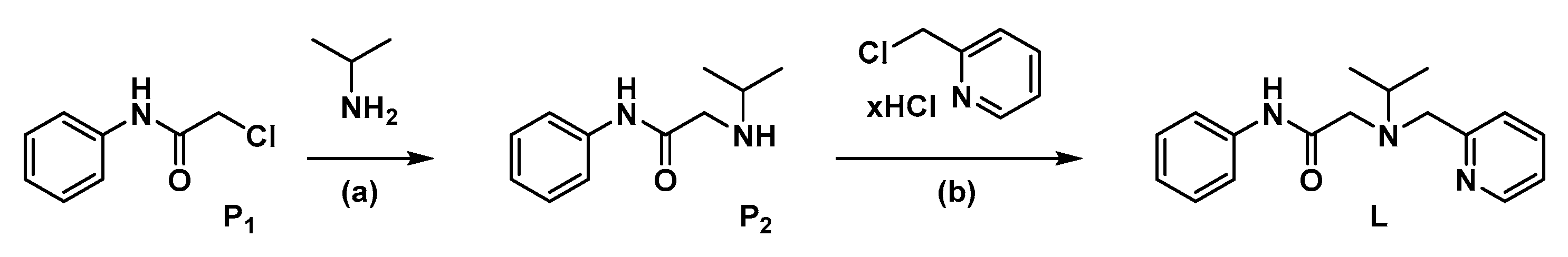
2.1.2. Synthesis of P2
2.1.3. Synthesis of L
2.1.4. Preparation of the [Zn(L2)]2+ Complex for NMR Measurements
2.1.5. Synthesis of [Zn(L)2]2][SiF6][BF4]2
2.2. Calculation of the NMR Parameters
2.2.1. General Remarks
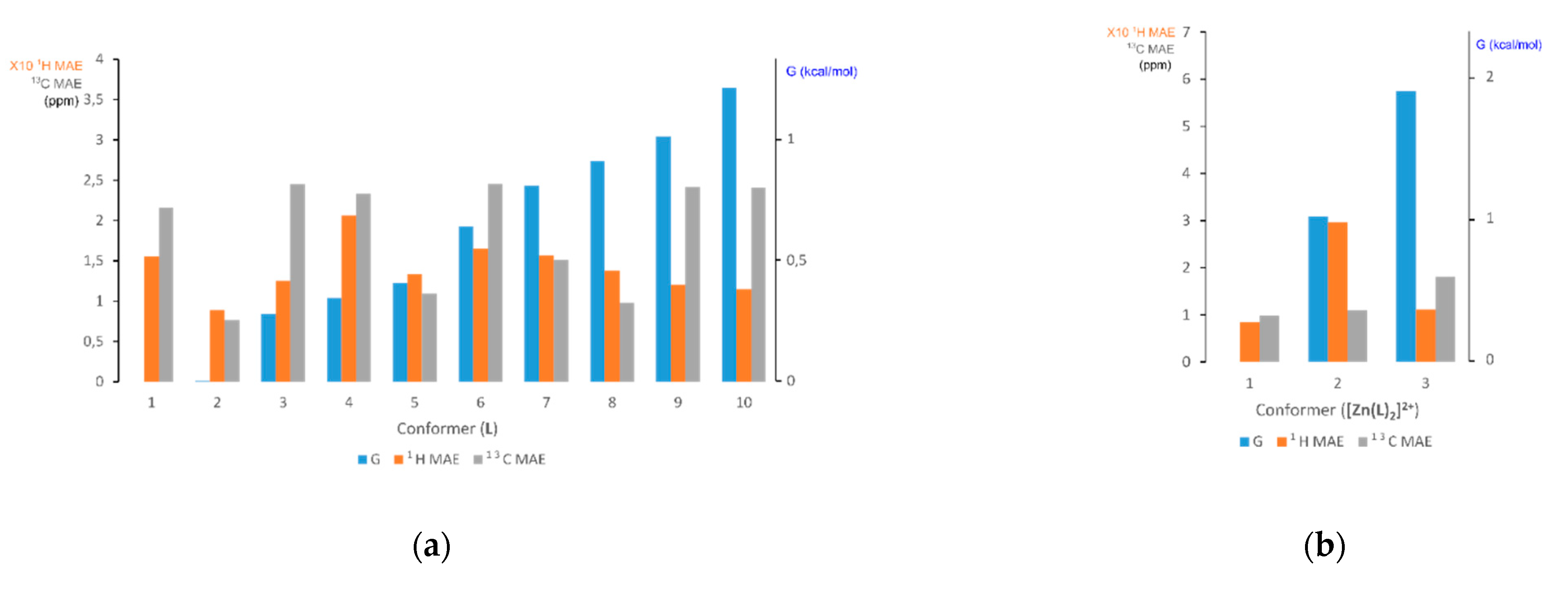
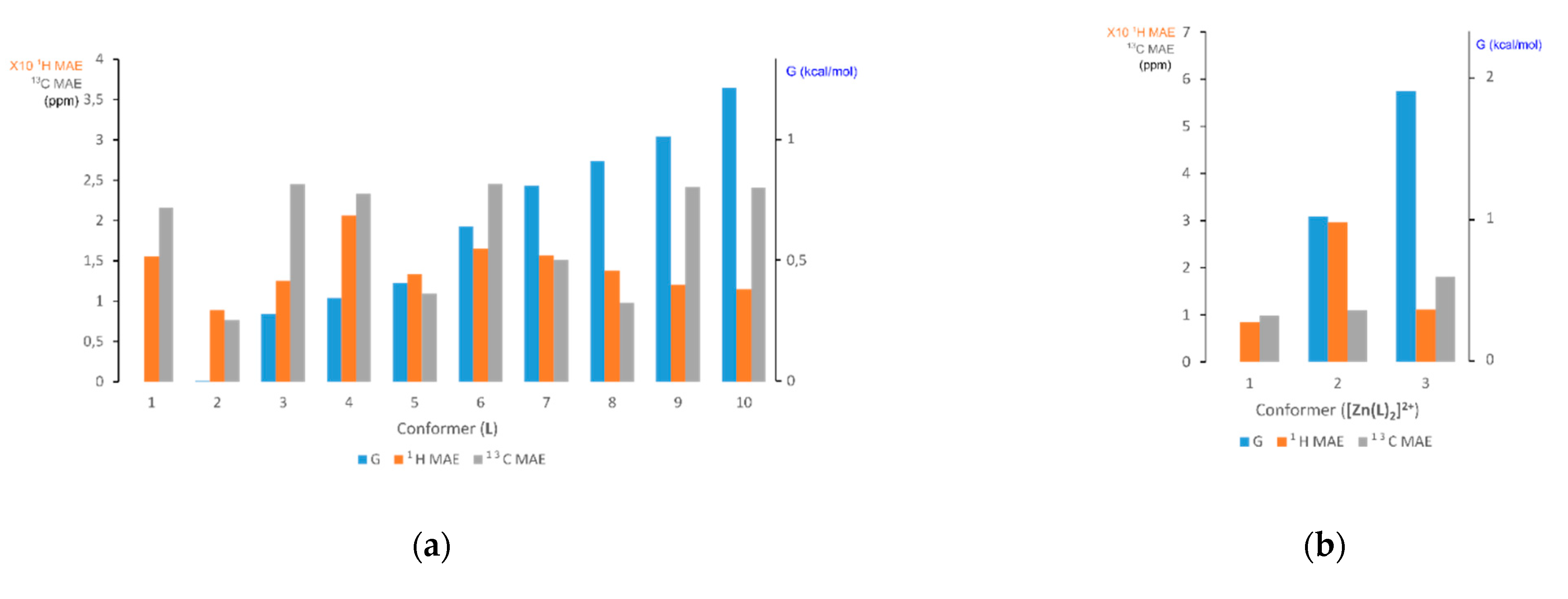
2.2.2. Generation of Conformer/Rotamer Ensembles (Step 1)
2.2.3. Free Energy Ranking of Conformers in Solution (Step 2)
2.2.4. Computation of NMR Parameters (Step 3)
2.2.5. Computation of the Averaged NMR Parameters and MAE Values (Step 4)
2.3. X-ray Diffraction
3. Results
3.1. Synthesis
3.2. NMR Analysis
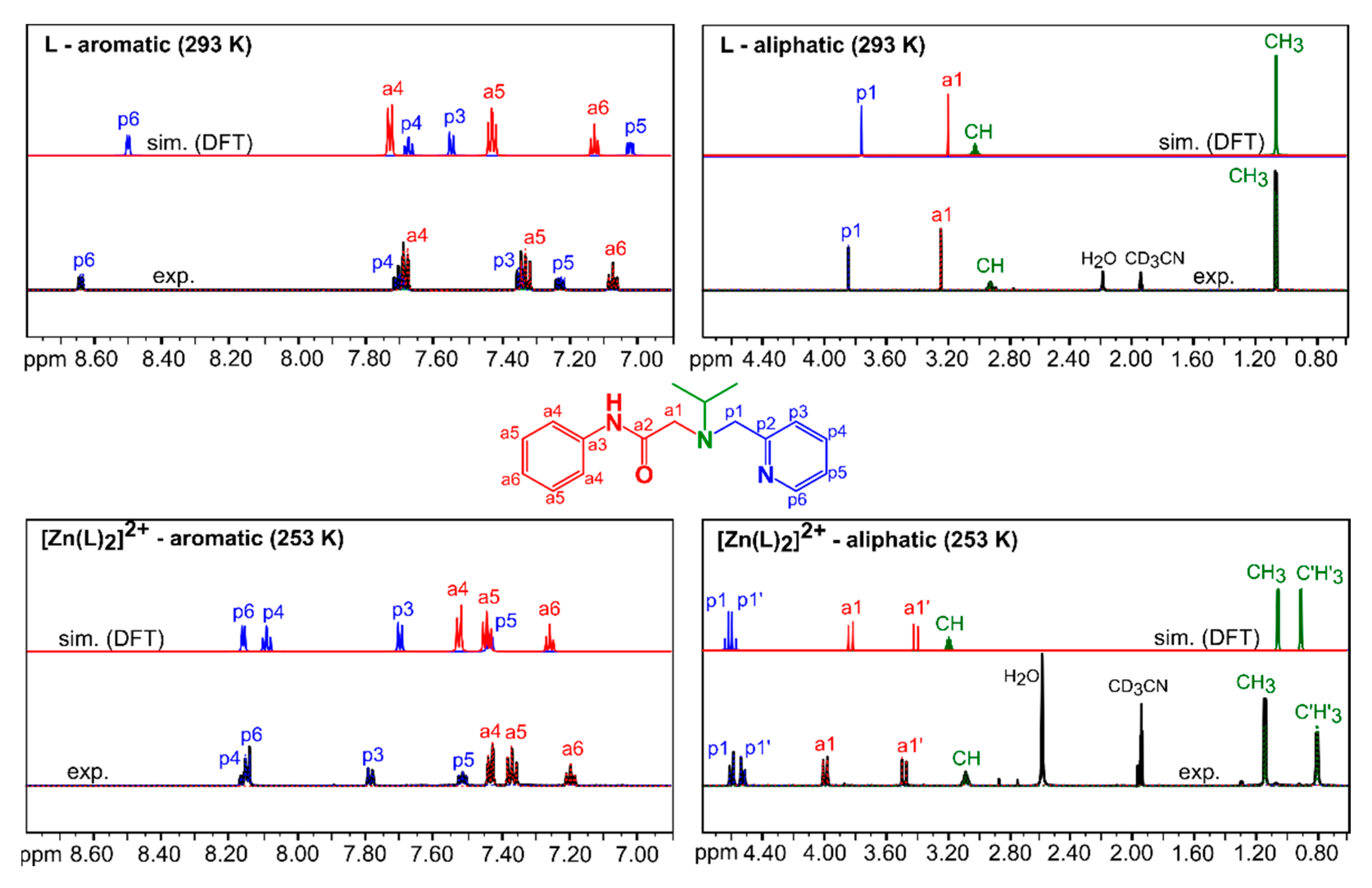
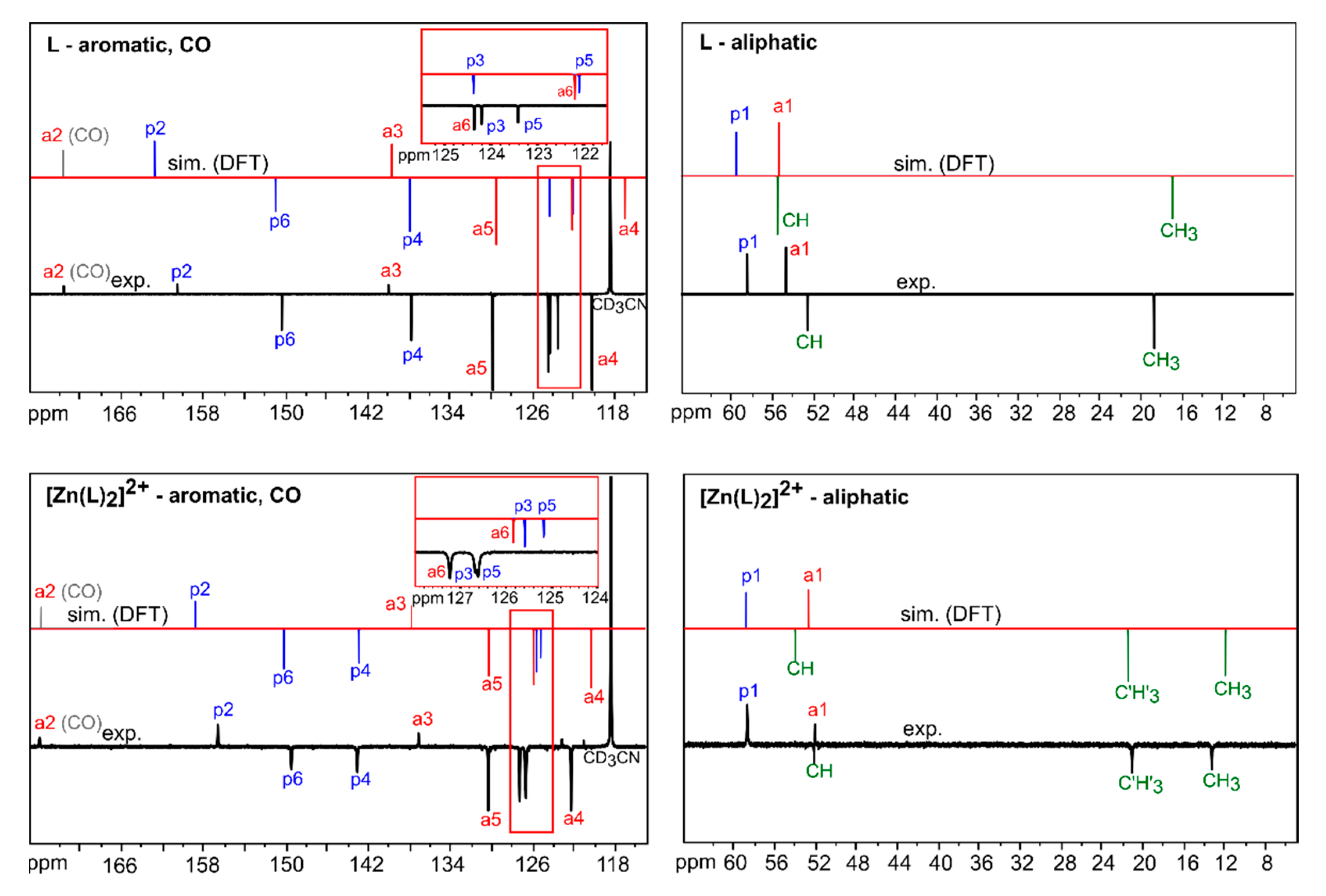
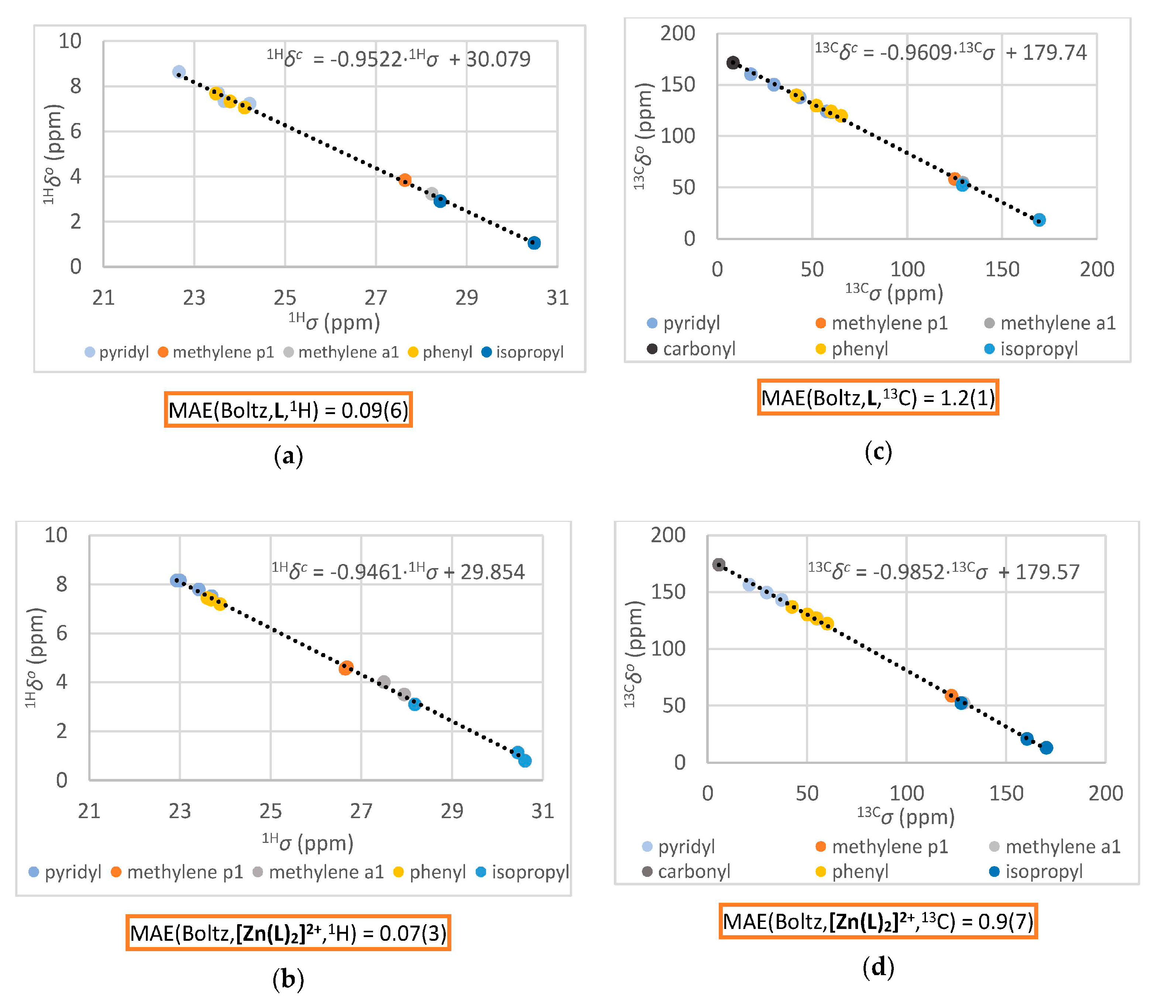
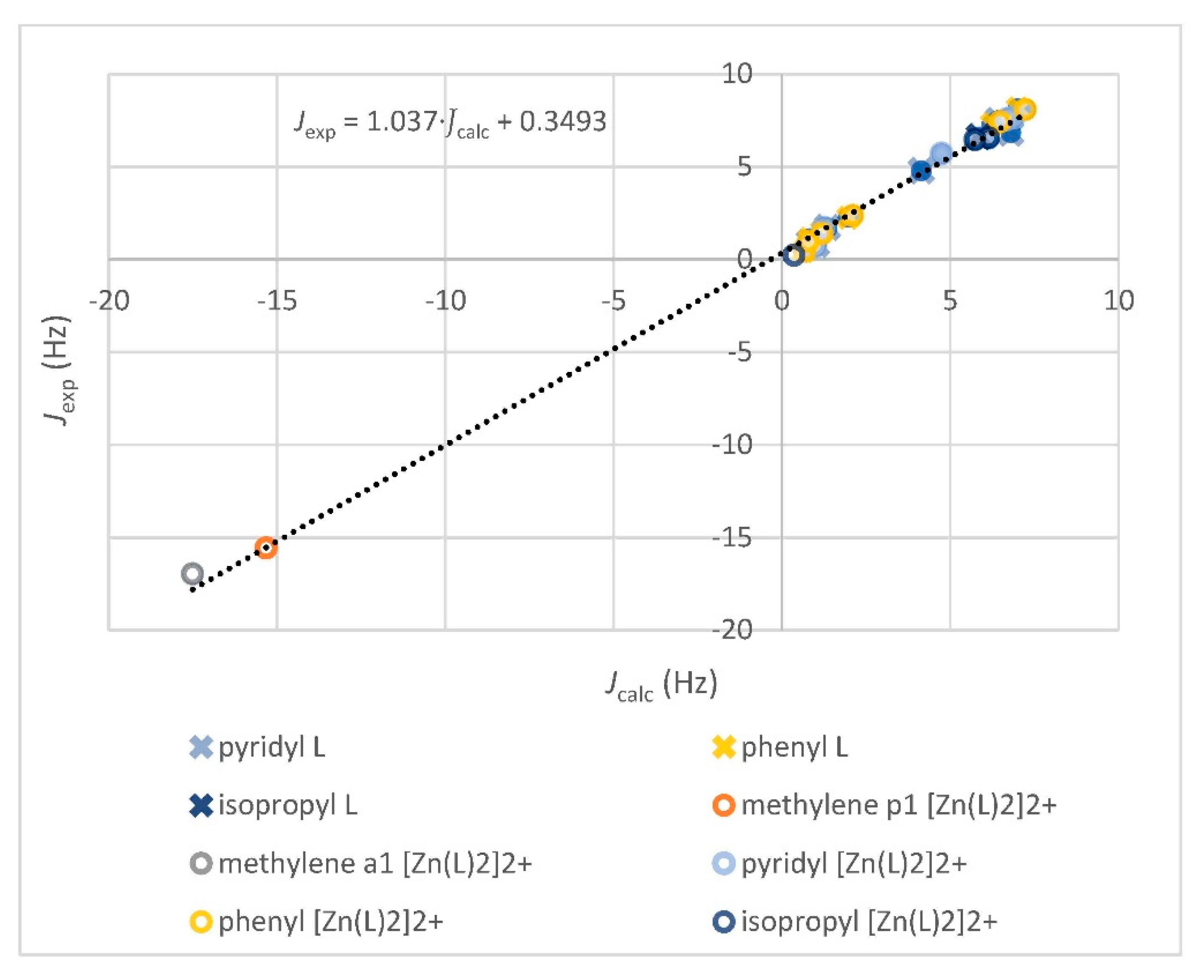
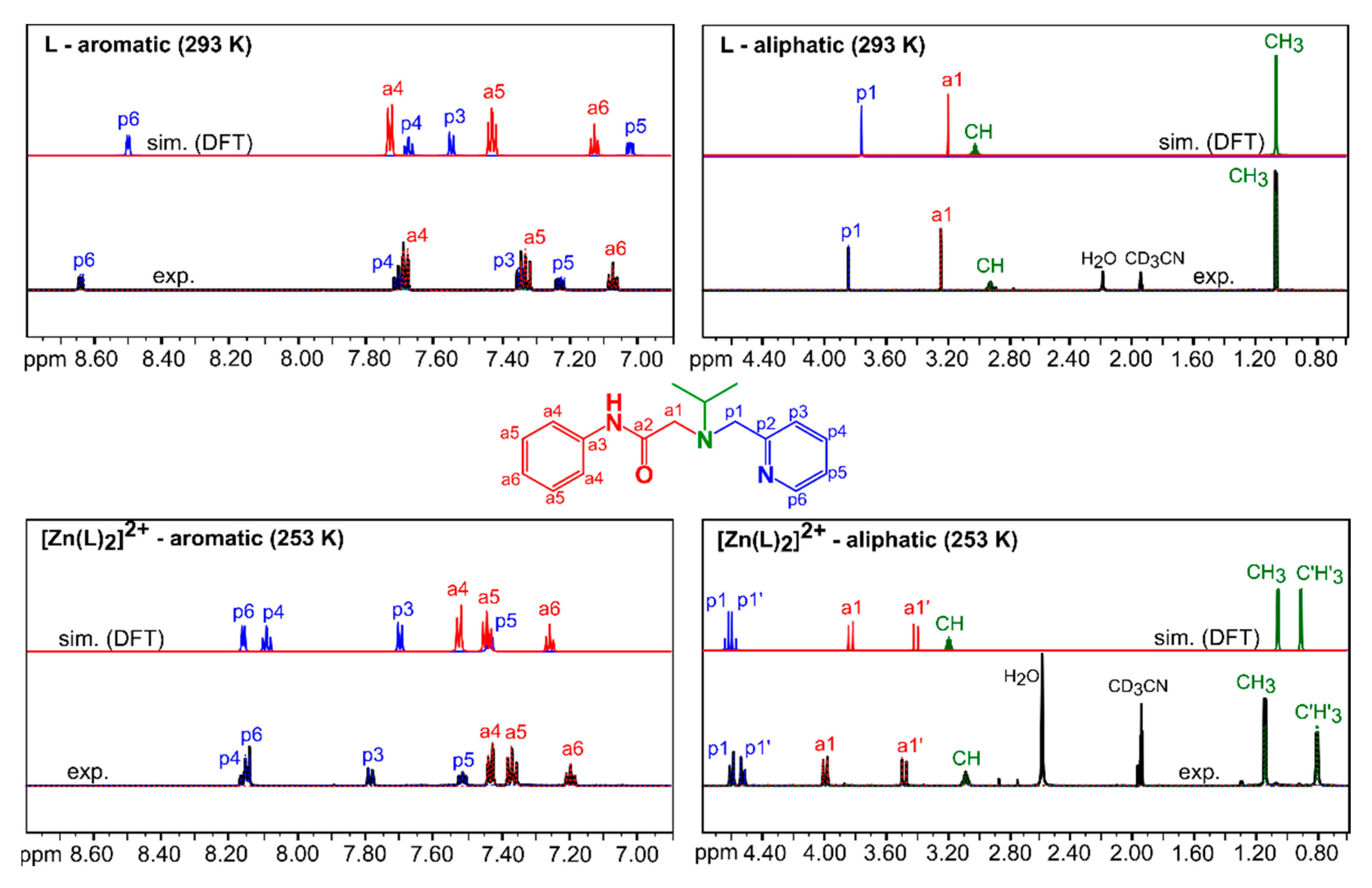
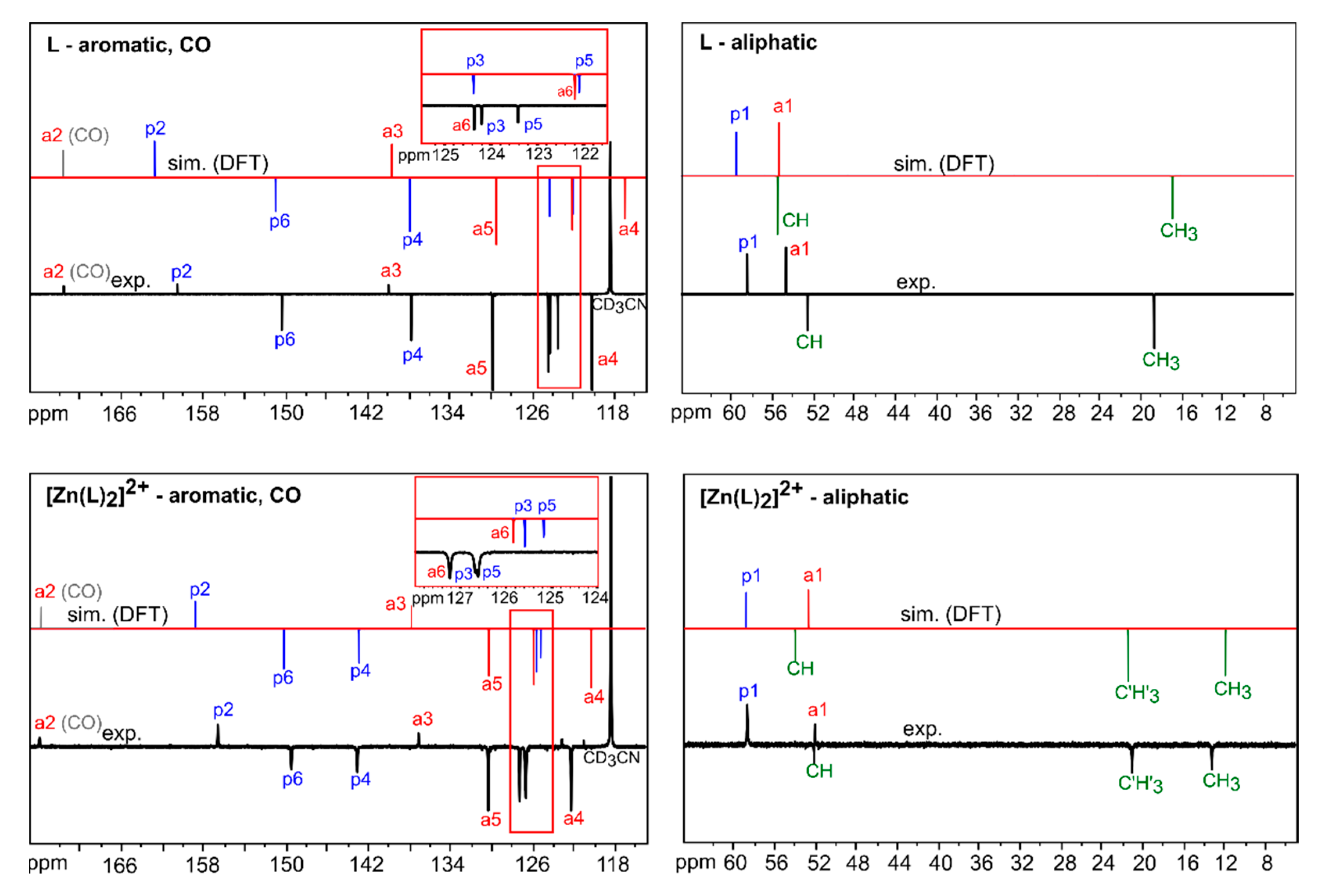
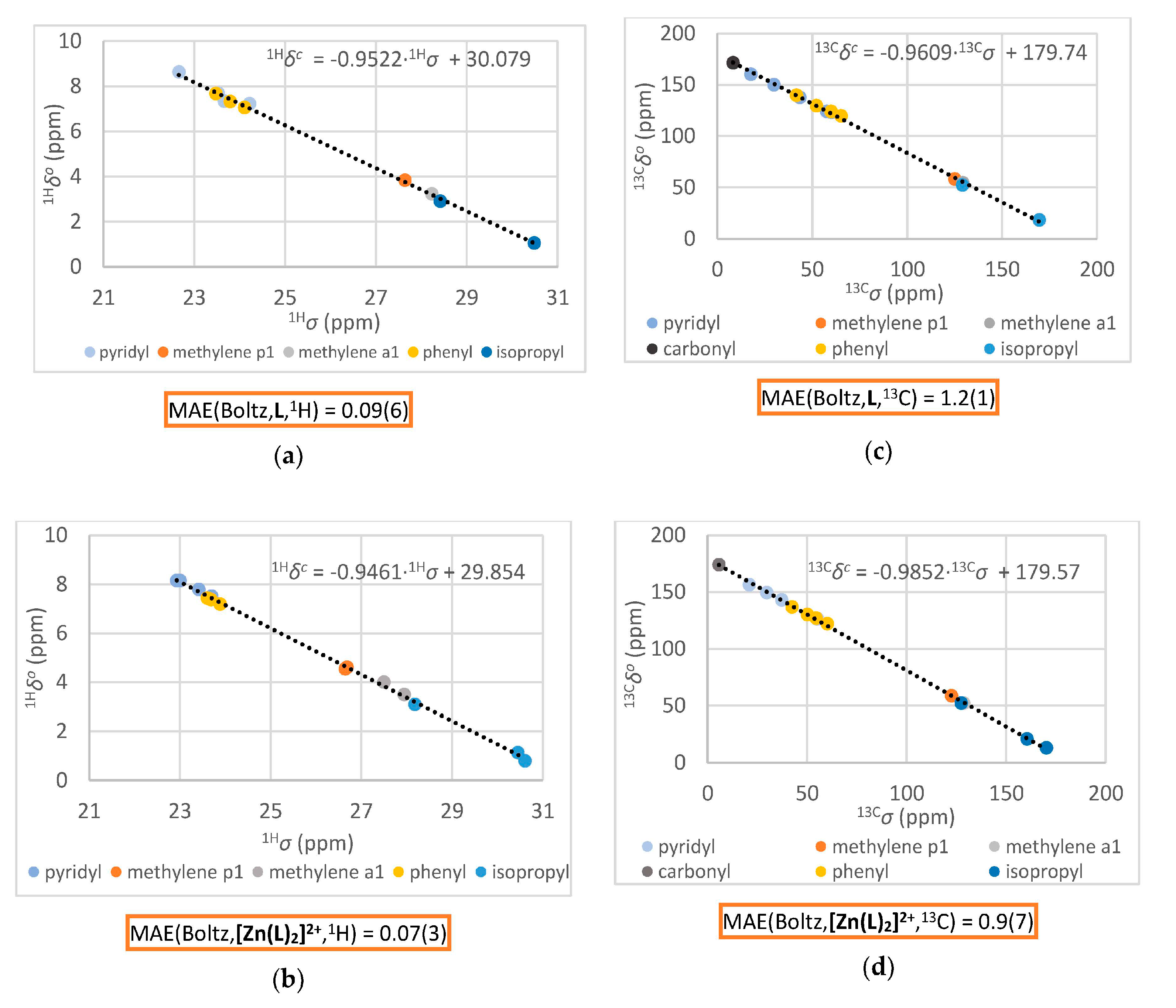
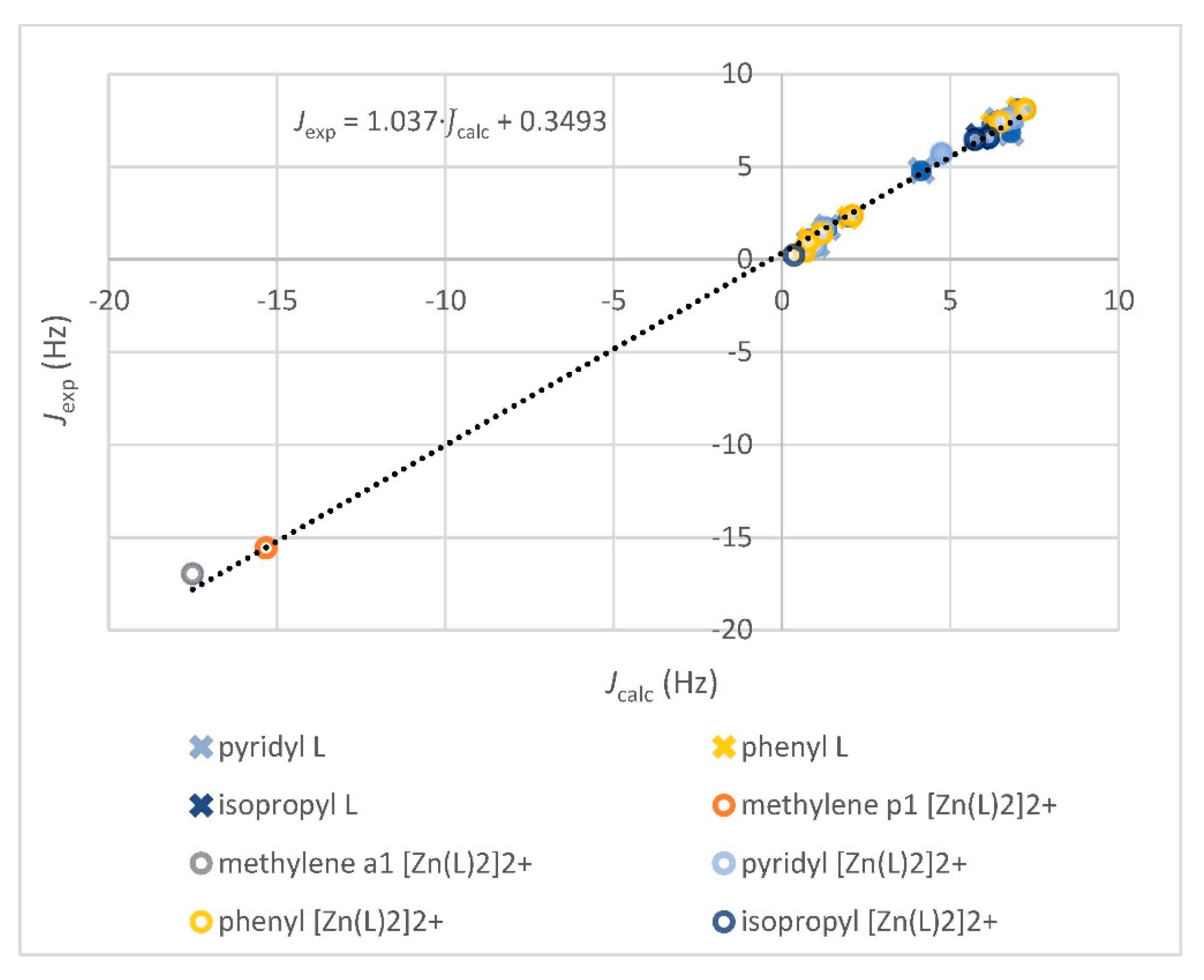
3.2.1. Comparison of Experimental and Simulated NMR Spectra
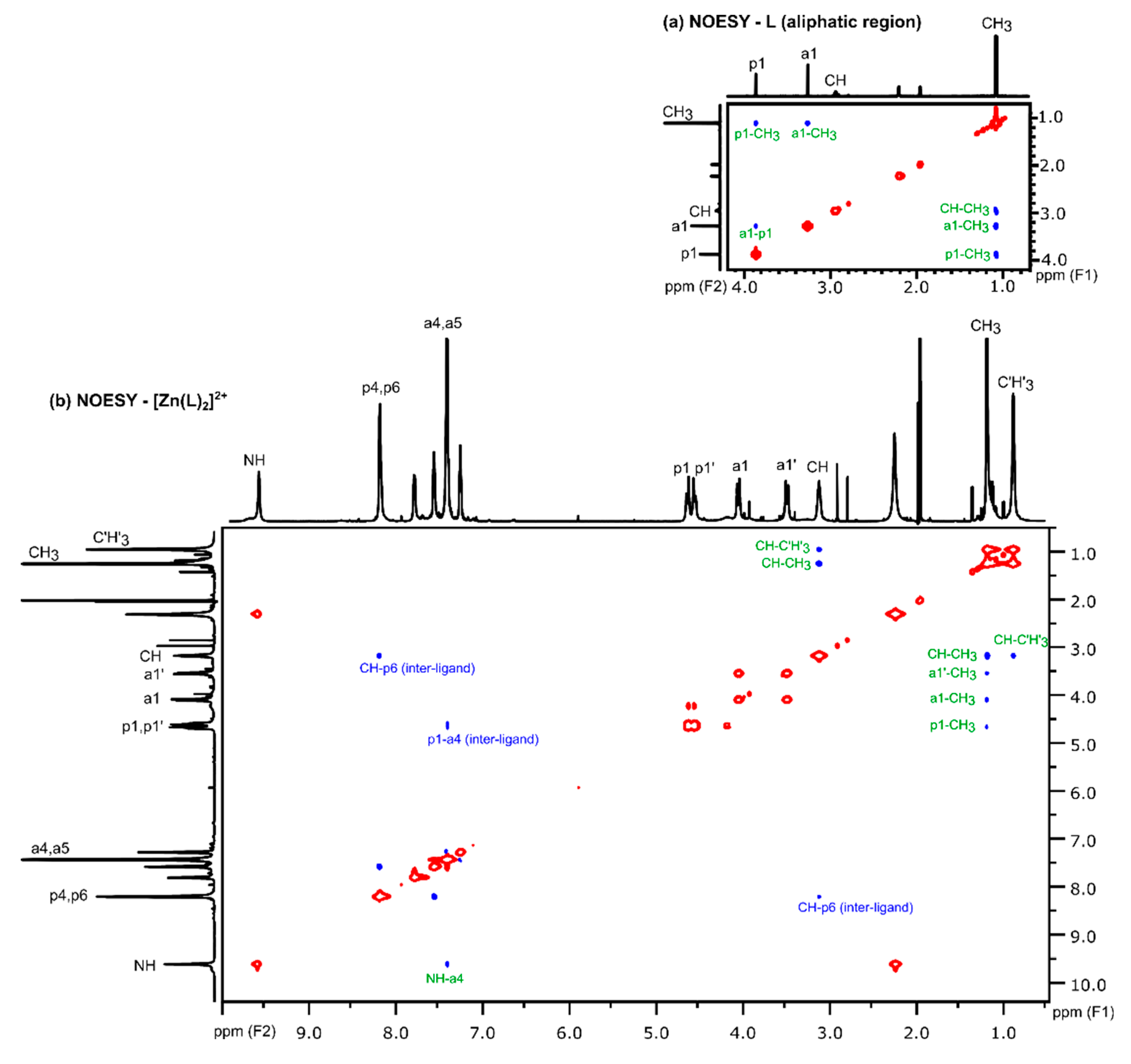
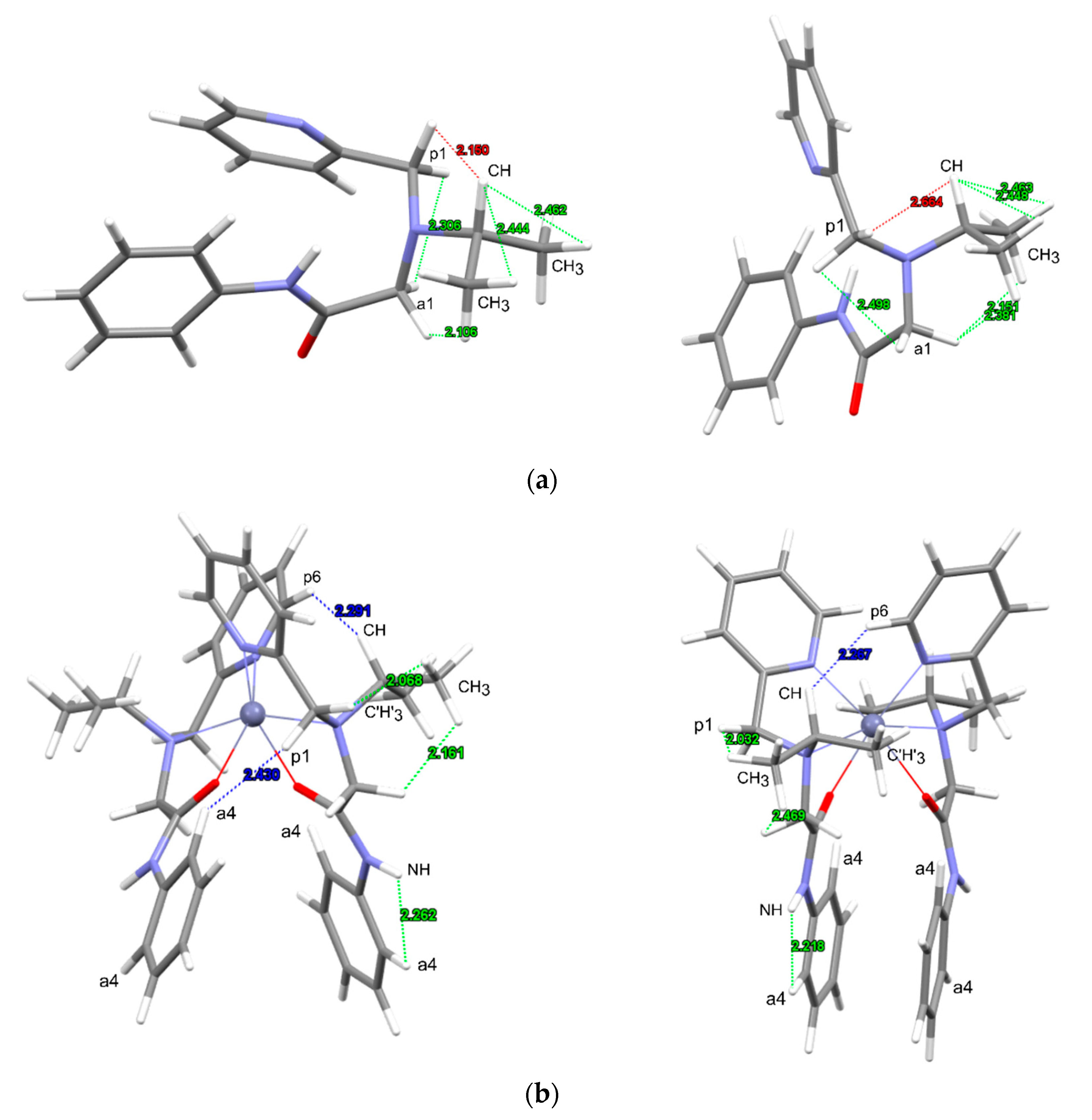
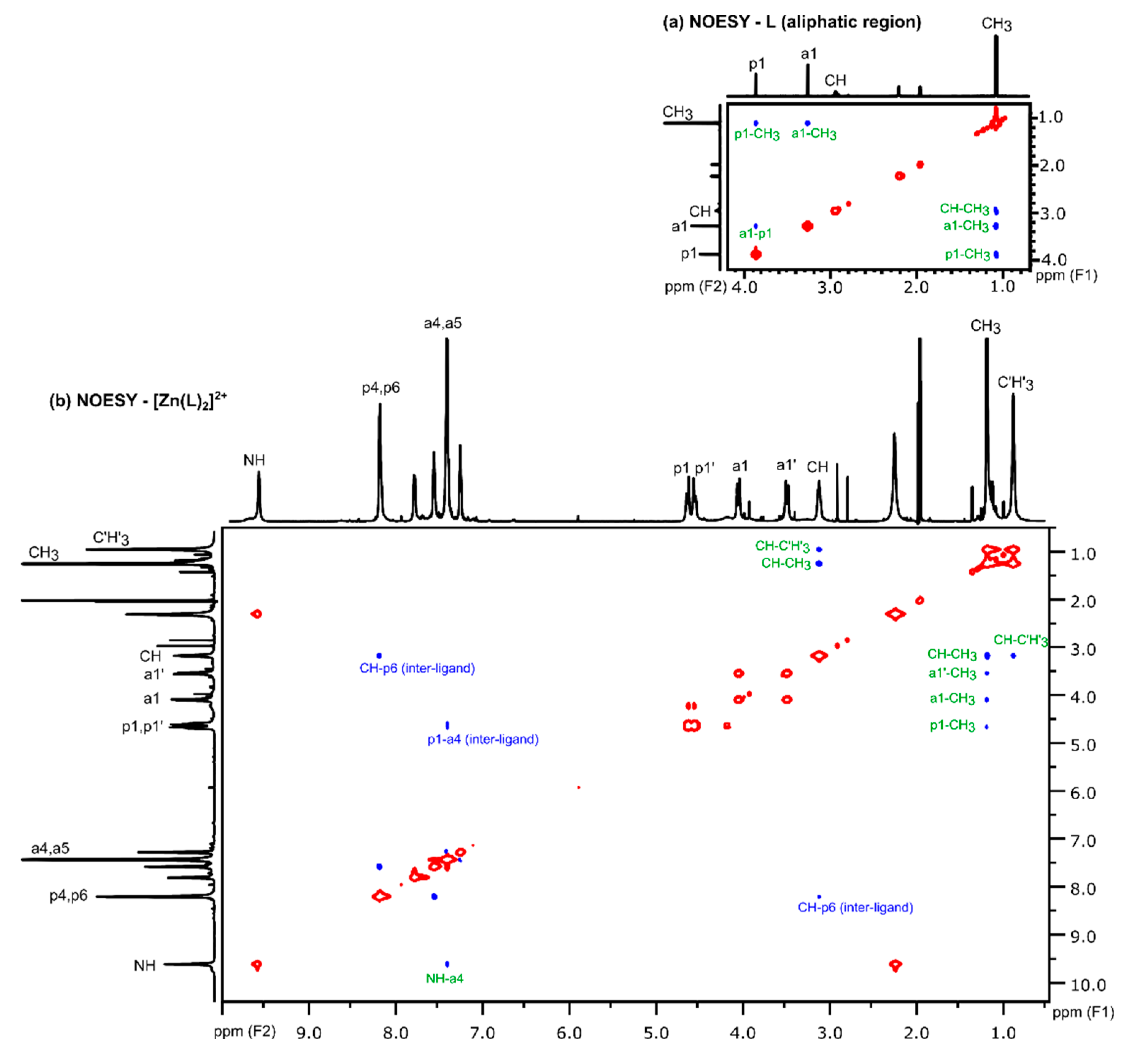
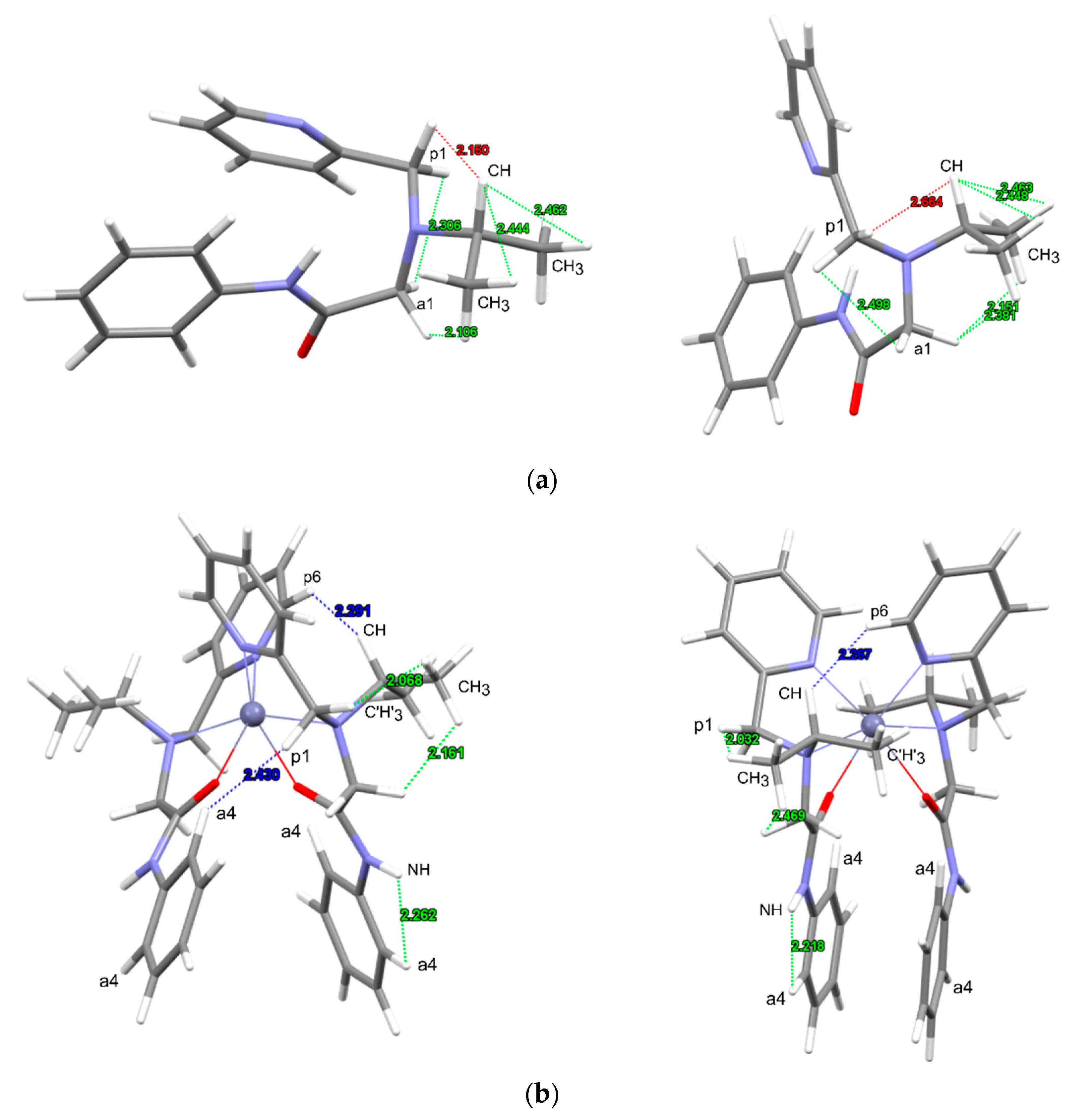
3.2.2. Analysis of NOESY Spectra
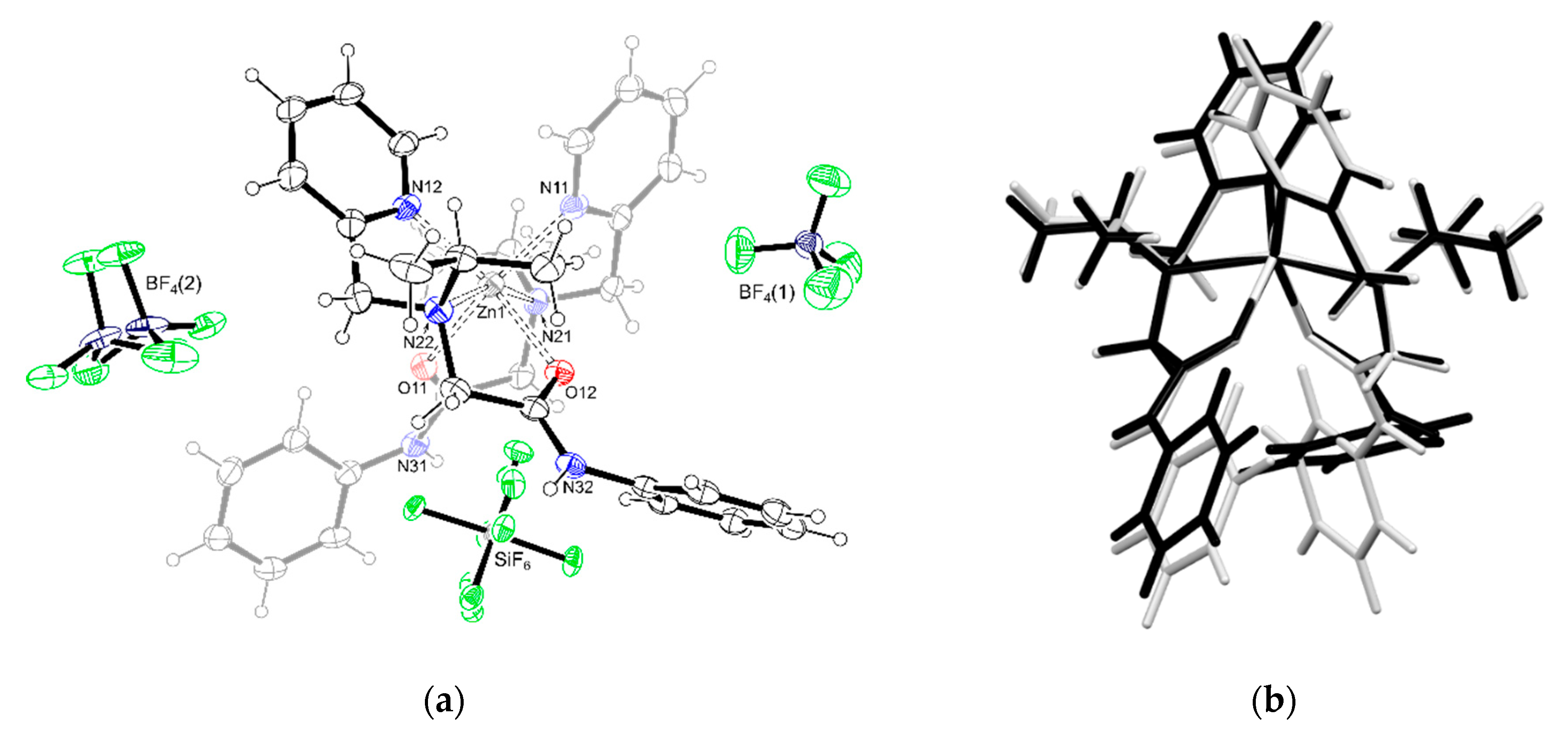
3.3. Crystal Structure of [Zn(L)2]2][SiF6][BF4]2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Willoughby, P.H.; Jansma, M.J.; Hoye, T.R. A guide to small-molecule structure assignment through computation of (1H and 13C) NMR chemical shifts. Nat. Protoc. 2014, 9, 643–660. [Google Scholar] [CrossRef] [PubMed]
- Viterbo, D. Solution and refinement of crystal structures. In Fundamentals of Crystallography, 2nd ed.; Giacovazzo, C., Ed.; Oxford University Press: Oxford, UK, 2002; p. 414. [Google Scholar]
- Pantalon Juraj, N.; Kirin, S.I. Inorganic stereochemistry: Geometric isomerism in bis-tridentate ligand. Coord. Chem. Rev. 2021, 445, 214051. [Google Scholar] [CrossRef]
- Pantalon Juraj, N.; Krklec, M.; Novosel, T.; Perić, B.; Vianello, R.; Raić-Malić, S.; Kirin, S.I. Copper(II) and zinc(II) complexes of mono- and bis-1,2,3-triazole-substituted heterocyclic ligands. Dalton Trans. 2020, 49, 9002–9015. [Google Scholar] [CrossRef]
- Pantalon Juraj, N.; Miletić, G.I.; Perić, B.; Popović, Z.; Smrečki, N.; Vianello, R.; Kirin, S.I. Stereochemistry of Hexacoordinated Zn(II), Cu(II), Ni(II), and Co(II) Complexes with Iminodiacetamide Ligands. Inorg. Chem. 2019, 58, 16445–16457. [Google Scholar] [CrossRef] [PubMed]
- Pantalon Juraj, N.; Muratović, S.; Perić, B.; Šijaković Vujičić, N.; Vianello, R.; Žilić, D.; Jagličić, Z.; Kirin, S.I. Structural Variety of Isopropyl-bis(2-picolyl)amine Complexes with Zinc(II) and Copper(II). Cryst. Growth Des. 2020, 20, 2440–2453. [Google Scholar] [CrossRef]
- Pantalon Juraj, N.; Tandarić, T.; Tadić, V.; Perić, B.; Moreth, D.; Schatzschneider, U.; Brozovic, A.; Vianello, R.; Kirin, S.I. Tuning the coordination properties of chiral pseudopeptide bis(2-picolyl)amine and iminodiacetamide ligands in Zn(ii) and Cu(ii) complexes. Dalton Trans. 2022, 51, 17008–17021. [Google Scholar] [CrossRef]
- Grimme, S.; Bannwarth, C.; Dohm, S.; Hansen, A.; Pisarek, J.; Pracht, P.; Seibert, J.; Neese, F. Fully Automated Quantum-Chemistry-Based Computation of Spin–Spin-Coupled Nuclear Magnetic Resonance Spectra. Angew. Chem. Int. Ed. 2017, 56, 14763–14769. [Google Scholar] [CrossRef]
- Pantalon Juraj, N.; Le Pennec, J.; Perić, B.; Kirin, S.I. A Case Study of Supramolecular Organization: One Ferrocene Substituted Iminodiacetamide and Its Chloroform Solvate. Croat. Chem. Acta 2017, 90, 613–623. [Google Scholar]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.S.; Quirt, A.R. NMR Spectra of Symmetric Molecules. I. The spin Hamiltonian for Twofold Symmetry. J. Magn. Reson. 1971, 5, 318–327. [Google Scholar]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Grimme, S.; Hansen, A.; Ehlert, S.; Mewes, J.-M. r2SCAN-3c: A “Swiss army knife” composite electronic-structure method. J. Chem. Phys. 2021, 154, 064103. [Google Scholar] [CrossRef] [PubMed]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Exploration of chemical compound, conformer, and reaction space with meta-dynamics simulations based on tight-binding quantum chemical calculations. J. Chem. Theory Comput. 2019, 15, 2847–2862. [Google Scholar] [CrossRef]
- Grimme, S.; Bannwarth, C.; Shushkov, P. A Robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent interactions of large molecular systems parametrized for all spd-block elements (Z = 1–86). J. Chem. Theory Comput. 2017, 13, 1989–2009. [Google Scholar] [CrossRef]
- Grimme, S.; Bohle, F.; Hansen, A.; Pracht, P.; Spicher, S.; Stahn, M. Efficient quantum chemical calculation of structure ensembles and free energies for nonrigid molecules. J. Phys. Chem. A 2021, 125, 4039–4054. [Google Scholar] [CrossRef]
- Bruker SAINT (v8.40A); Bruker AXS Inc.: Madison, WI, USA, 2019.
- Bruker SADABS (v2014/4); Bruker AXS Inc.: Madison, WI, USA, 2014.
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Aroyo, M.I.; Perez-Mato, J.M.; Capillas, C.; Kroumova, E.; Ivantchev, S.; Madariaga, G.; Kirov, A.; Wondratschek, H. Bilbao Crystallographic Server I: Databases and crystallographic computing programs. Z. Krist. 2006, 221, 15–27. [Google Scholar] [CrossRef]
- Guzei, I.A. An idealized molecular geometry library for refinement of poorly behaved molecular fragments with constraints. J. Appl. Crystallogr. 2014, 47, 806–809. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Cryst. C 2015, 71, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Müller, E.; Bayer, O.; Meerwein, H.; Ziegler, K. Band XI/2–Stickstoffverbindungen II Und III. In Methoden der Organischen Chemie (Houben Weyl), Band XI/2–Stickstoffverbindungen II und III; Georg Thieme Verlag: Stuttgart, Germany, 1958. [Google Scholar]
- Wen, Y.; Chen, X.; Wen, H.; Tang, X. Efficient Synthesis of Piperazinediones Using Potassium Iodide Catalysis in Aqueous Media. Lett. Org. Chem. 2011, 8, 732–736. [Google Scholar] [CrossRef]
- Saihara, K.; Yoshimura, Y.; Fujimoto, H.; Shimizu, A. Detrimental Effect of Glass Sample Tubes on Investigations of BF4--Based Room Temperature Ionic Liquid-Water Mixtures. J. Mol. Liq. 2016, 219, 493–496. [Google Scholar] [CrossRef]
- Casellas, H.; Pevec, A.; Kozlevcar, B.; Gamez, P.; Reedijk, J. An Unprecedented M4-SiF62−- Bridged Supramolecular Polymer Consisting of Bis-μ-F-Bridged Dinuclear Cu(II) Dications. Polyhedron 2005, 24, 1549–1554. [Google Scholar] [CrossRef]
- Levitt, M.H. Spin Dynamics: Basis of Nuclear Magnetic Resonance; John Wiley & Sons, Ltd.: Chichester, UK, 2001; pp. 479–511. [Google Scholar]
- Ruden, T.A.; Ruud, K. Ro-Vibrational Corrections to NMR Parameters. In Calculation of NMR and EPR Parameters, Theory and Applications; Kaupp, M., Bühl, M., Malkin, V.G., Eds.; WILEY-VCH: Weinheim, Germany, 2004; pp. 153–173. [Google Scholar]
- Faruggia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Sternberg, U.; Witter, R.; Ulrich, A.S. Crystal Structure Refinement Using Chemical Shifts. In Modern Magnetic Resonance; Webb, G.A., Ed.; Springer: Dordrecht, The Netherlands, 2008; pp. 71–78. [Google Scholar]
- Jakovkin, I.; Klipfel, M.; Muhle-Goll, C.; Ulrich, A.S.; Luy, B.; Sternberg, U. Rapid calculation of protein chemical shifts using bond polarization theory and its application to protein structure refinement. Phys. Chem. Chem. Phys. 2012, 14, 12263–12276. [Google Scholar] [CrossRef]
- Prieß, W.; Sternberg, U. Fast calculation of 13C NMR chemical shift tensors using the bond polarization model. J. Mol. Struct. 2001, 544, 181–190. [Google Scholar] [CrossRef]
- Opačak, S.; Perić, B.; Gojšić, T.; Čikoš, A.; Vikić-Topić, D.; Kirin, S.I. Tandem amide coupling and hydroamination: Unexpected benzotriazole oxide addition to the propiolic acid triple bond. New J. Chem. 2022, 46, 13275–13285. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula | [Zn(L)2]2][SiF6][BrF4]2 |
|---|---|
| Fw (g mol−1) | 1579.92 |
| Crystal system | cubic |
| Space group | (No. 222) |
| a (Å) | 37.0820(3) |
| b (Å) | 37.0820(3) |
| c (Å) | 37.0820(3) |
| α (o) | 90.0 |
| β (o) | 90.0 |
| γ (o) | 90.0 |
| V (Å3) | 50,990.5(12) |
| Z | 24 |
| Dcalc (g cm−3) | 1.235 |
| F(000) | 19632 |
| Temperature (K) | 100 |
| Reflections collected | 157629 |
| Independent reflections | 8722 |
| Rinit | 0.0304 |
| Reflections observed [I > 2σ(I)] | 7823 |
| Parameters | 501 |
| R1 1 | 0.0599 |
| wR2 (all data) 2 | 0.1833 |
| Goof, S 3 | 1.061 |
| Maximum/minimumelectron density (e Å3) | 1.090/−0.580 |
| Spin System | Parameter | L Observed (293 K) | L Calculated (293 K) 1 | [Zn(L)2]2+ Observed (253 K) | [Zn(L)2]2+ Calculated (253 K) 1 |
|---|---|---|---|---|---|
| pyridyl | 1Hδ(p3) | 7.35(1) | 7.548 (23.6620) | 7.79(1) | 7.698 (23.4181) |
| 1Hδ(p4) | 7.70(1) | 7.674 (23.5292) | 8.15(1) | 8.091 (23.0032) | |
| 1Hδ(p5) | 7.23(1) | 7.020 (24.2167) | 7.51(1) | 7.434 (23.6978) | |
| 1Hδ(p6) | 8.64(1) | 8.502 (22.6604) | 8.15(1) | 8.160 (22.9295) | |
| 3J(p3,p4) | 6.84(1) | 6.79 | 7.66(1) | 6.87 | |
| 3J(p4,p5) | 7.50(1) | 6.41 | 7.57(1) | 6.67 | |
| 3J(p5,p6) | 4.80(1) | 4.13 | 5.71(1) | 4.73 | |
| 4J(p3,p5) 2 | 0.9(1) | 0.81 | 0.8(1) | 0.91 | |
| 4J(p4,p6) 2 | 1.7(1) | 1.34 | 1.7(1) | 1.23 | |
| 5J(p3,p6) 2 | 0.8(1) | 1.02 | 0.7(1) | 0.99 | |
| phenyl | 1Hδ(a4) | 7.68(1) | 7.729 (23.4724) | 7.43(1) | 7.526 (23.6005) |
| 1Hδ(a5) | 7.33(1) | 7.428 (23.7882) | 7.37(1) | 7.444 (23.6859) | |
| 1Hδ(a6) | 7.07(1) | 7.126 (24.1051) | 7.19(1) | 7.259 (23.8827) | |
| 3J(a4,a5) | 8.16(1) | 7.00 | 8.10(1) | 7.23 | |
| 3J(a5,a6) | 7.31(1) | 6.23 | 7.43(1) | 6.47 | |
| 4J(a4,a4) 2 | 2.3(1) | 1.96 | 2.4(1) | 2.10 | |
| 4J(a5,a5) 2 | 1.5(1) | 1.19 | 1.4(1) | 1.19 | |
| 4J(a4,a6) 2 | 1.1(1) | 0.81 | 1.0(1) | 0.78 | |
| 5J(a4,a5) 2 | 0.5(1) | 0.64 | 0.4(1) | 0.66 | |
| methylene p1 | 1Hδ(p1) | 3.85(1) | 3.764 (27.6356) | 4.61(1) | 4.645 (26.6455) |
| 1Hδ(p1′) | 4.54(1) | 4.605 (26.6874) | |||
| 2J(p1,p1′) | −15.56(1) | −15.33 | |||
| methylene a1 | 1Hδ(a1) | 3.25(1) | 3.200 (28.2281) | 4.00(1) | 3.836 (27.5006) |
| 1Hδ(a1′) | 3.45(1) | 3.412 (27.9480) | |||
| 2J(a1,a1′) | −16.96(1) | −17.51 | |||
| isopropyl | 1Hδ(CH) | 2.92(1) | 3.023 (28.4140) | 3.08(1) | 3.191 (28.2825) |
| 1Hδ(CH3) | 1.06(1) | 1.056 (30.4803) | 1.13(1) | 1.045 (30.4505) | |
| 1Hδ(C’H’3) | 0.79(1) | 0.895 (30.6087) | |||
| 3J(CH,CH3) | 6.6(1) | 5.86 | 6.5(1) | 5.74 | |
| 3J(CH,C’H’3) | 6.6(1) | 6.13 | |||
| 4J(CH3,C’H’3) 2 | 0.2(1) | 0.36 |
| Atom/Signal | L Observed (13Cδo, ppm) | L Calculated (13Cδc, ppm) 1 | [Zn(L)2]2+ Observed (13Cδo, ppm) | [Zn(L)2]2+ Calculated (13Cδc, ppm) 1 |
|---|---|---|---|---|
| p1 | 58.54(1) | 59.63 (124.997) | 58.72(1) | 58.88 (122.501) |
| p2 | 160.54(1) | 162.81 (17.617) | 156.61(1) | 158.86 (21.019) |
| p3 | 124.16(1) | 124.36 (57.628) | 126.61(1) | 125.58 (54.801) |
| p4 | 137.71(1) | 138.03 (43.408) | 143.00(1) | 142.89 (37.234) |
| p5 | 123.37(1) | 122.08 (60.005) | 126.55(1) | 125.16 (55.224) |
| p6 | 150.33(1) | 151.13 (29.778) | 149.44(1) | 150.22 (29.792) |
| a1 | 54.72(1) | 55.49 (129.307) | 52.07(1) | 52.83 (128.643) |
| a2 | 171.67(1) | 171.75 (8.317) | 173.99(1) | 173.92 (5.733) |
| a3 | 139.91(1) | 139.75 (41.621) | 137.03(1) | 137.78 (42.419) |
| a4 | 120.07(1) | 117.03 (65.258) | 122.15(1) | 120.23 (60.231) |
| a5 | 129.76(1) | 129.57 (52.195) | 130.23(1) | 130.23 (50.083) |
| a6 | 124.32(1) | 122.18 (59.901) | 127.17(1) | 125.83 (54.551) |
| CH | 52.59(1) | 55.60 (129.191) | 52.14(1) | 54.06 (127.395) |
| CH3 | 18.58(1) | 16.85 (169.517) | 13.10(1) | 11.83 (170.257) |
| C’H’3 | 20.94(1) | 21.39 (160.552) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perić, B.; Pantalon Juraj, N.; Štefanić, Z.; Kirin, S.I. Structural Determination of the Hexacoordinated [Zn(L)2]2+ Complex Isomer Type Using Solution-State NMR, DFT Calculations and X-ray Crystallography. Crystals 2023, 13, 16. https://doi.org/10.3390/cryst13010016
Perić B, Pantalon Juraj N, Štefanić Z, Kirin SI. Structural Determination of the Hexacoordinated [Zn(L)2]2+ Complex Isomer Type Using Solution-State NMR, DFT Calculations and X-ray Crystallography. Crystals. 2023; 13(1):16. https://doi.org/10.3390/cryst13010016
Chicago/Turabian StylePerić, Berislav, Natalija Pantalon Juraj, Zoran Štefanić, and Srećko I. Kirin. 2023. "Structural Determination of the Hexacoordinated [Zn(L)2]2+ Complex Isomer Type Using Solution-State NMR, DFT Calculations and X-ray Crystallography" Crystals 13, no. 1: 16. https://doi.org/10.3390/cryst13010016