
Case Study on the Reproducibility of Characterizing the Electrocatalytic Activity of La1−xSrxMnO3 in ORR
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Synthesis of the LSMOx
2.2. Physical Characterization of the LSMOx
2.3. Preparation of the Ink
2.4. Electrocatalytic Characterization of the LSMOx on ORR
3. Results and Discussion
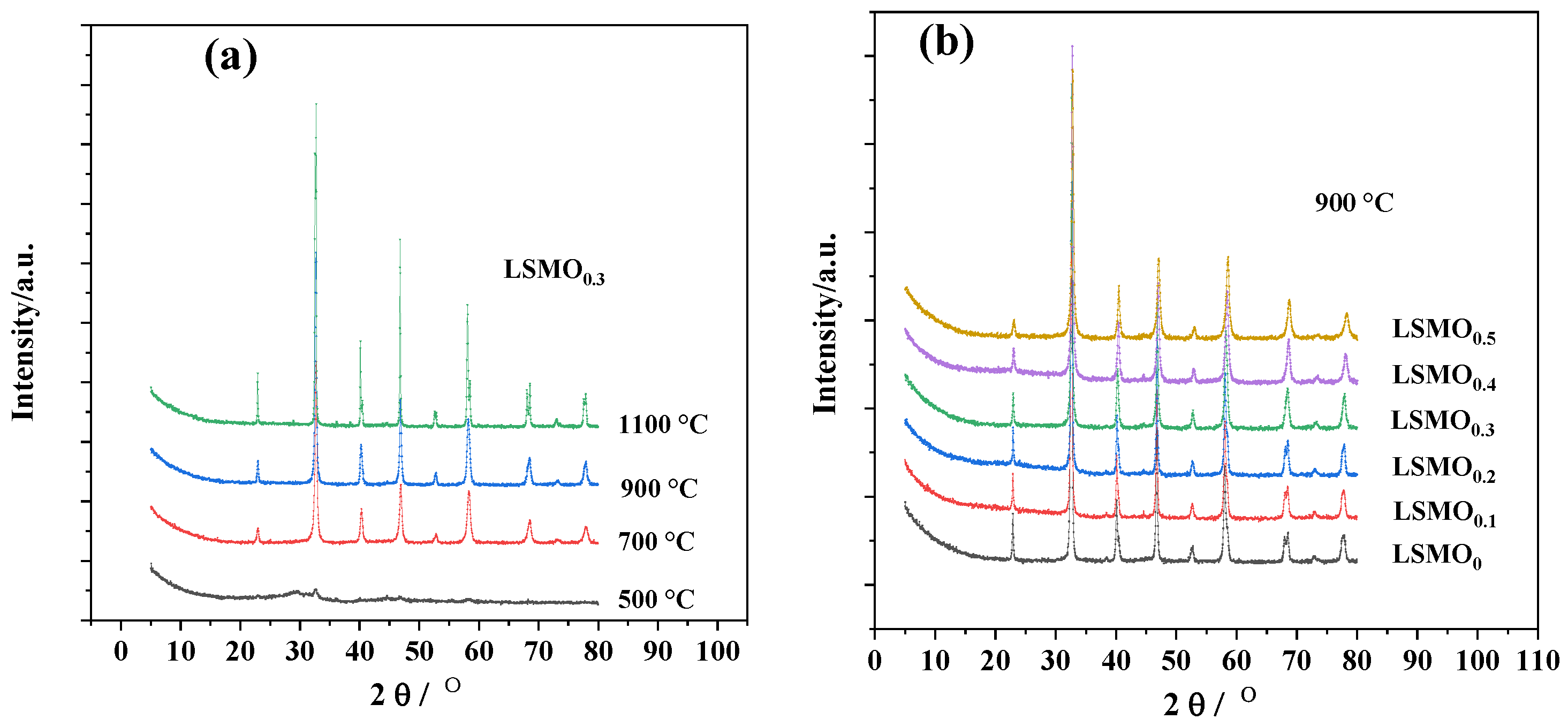
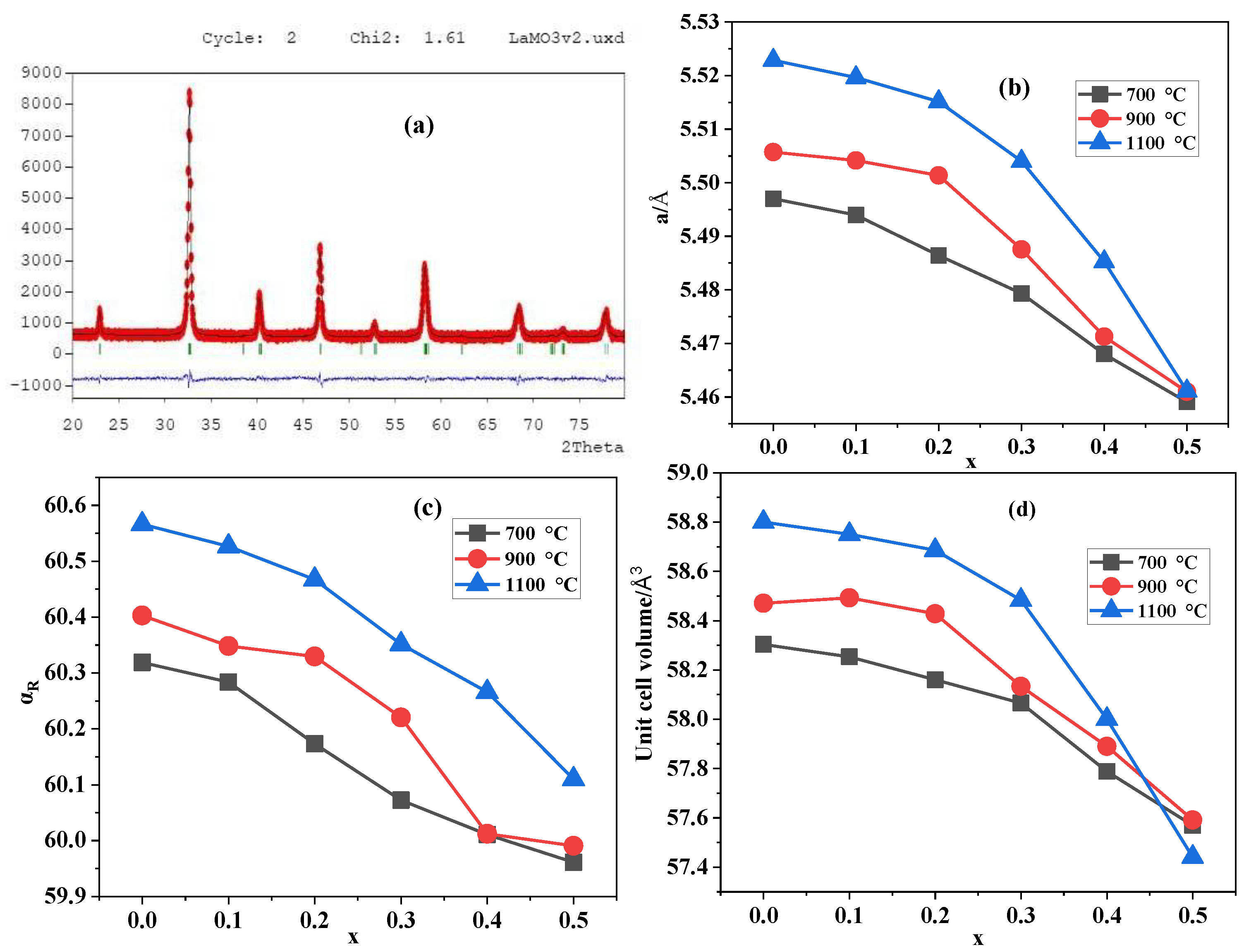
3.1. The Physical Characterization of the LSMOx
3.2. Reproducibility of the Electroactivity Characterization
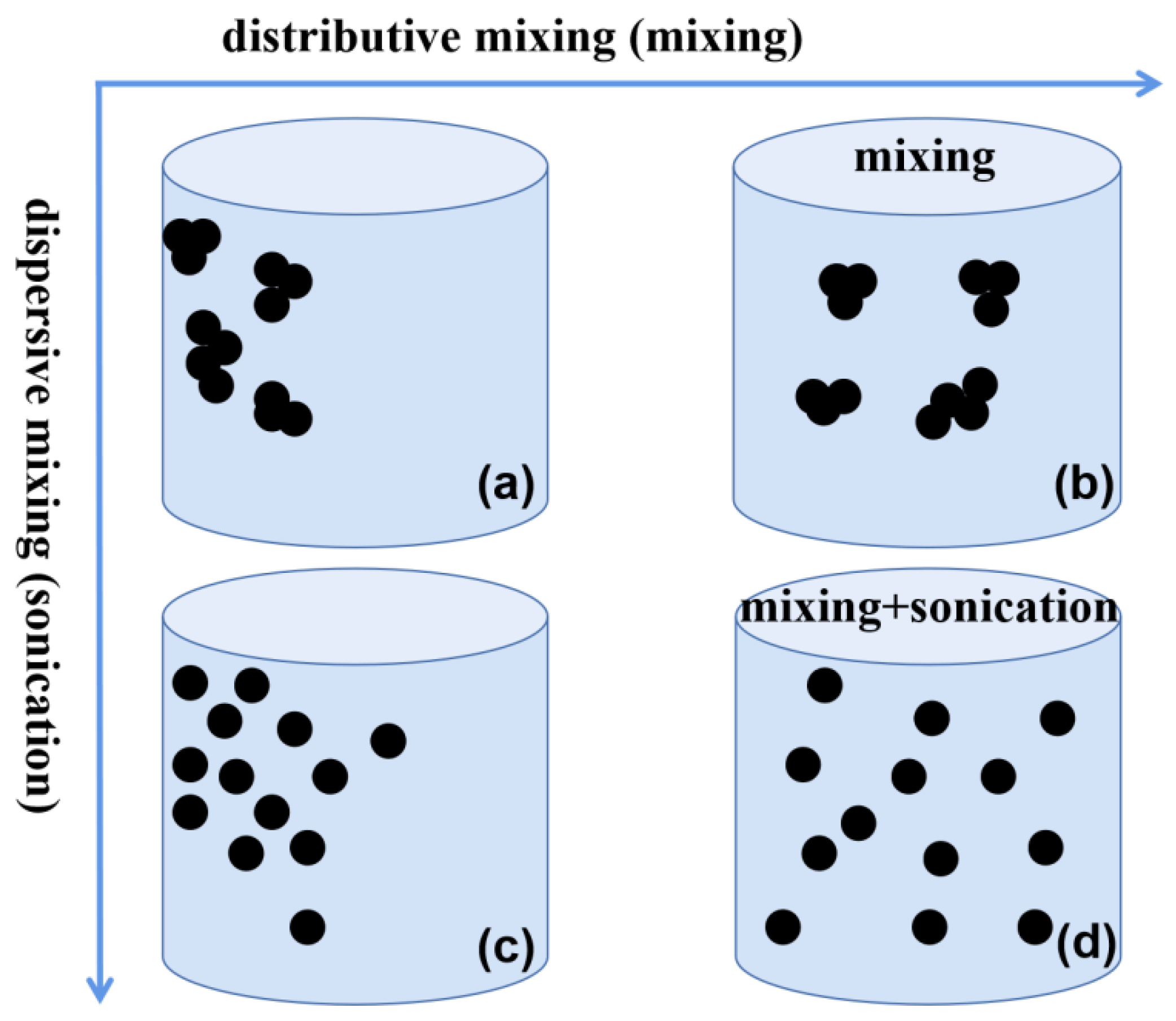
3.2.1. The Effect of the Manner of Mixing Ink
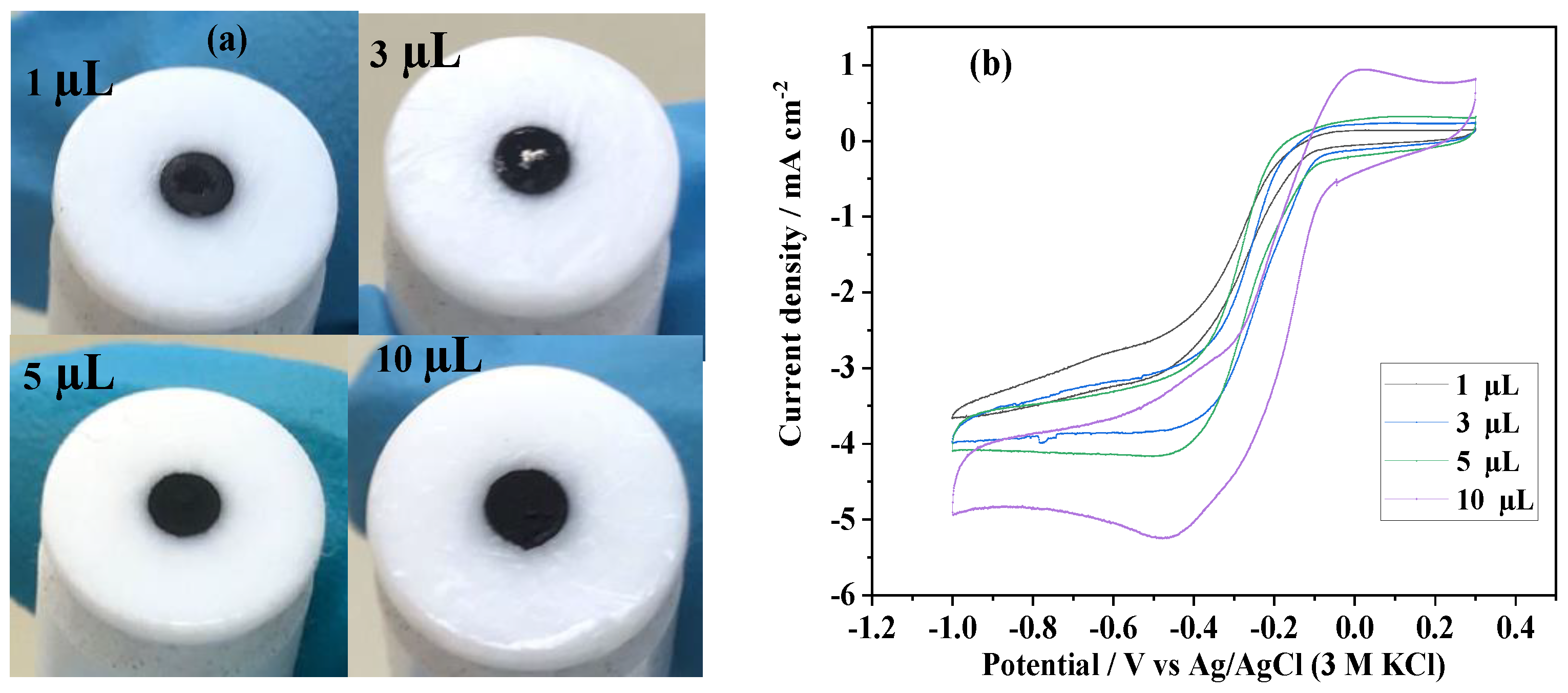
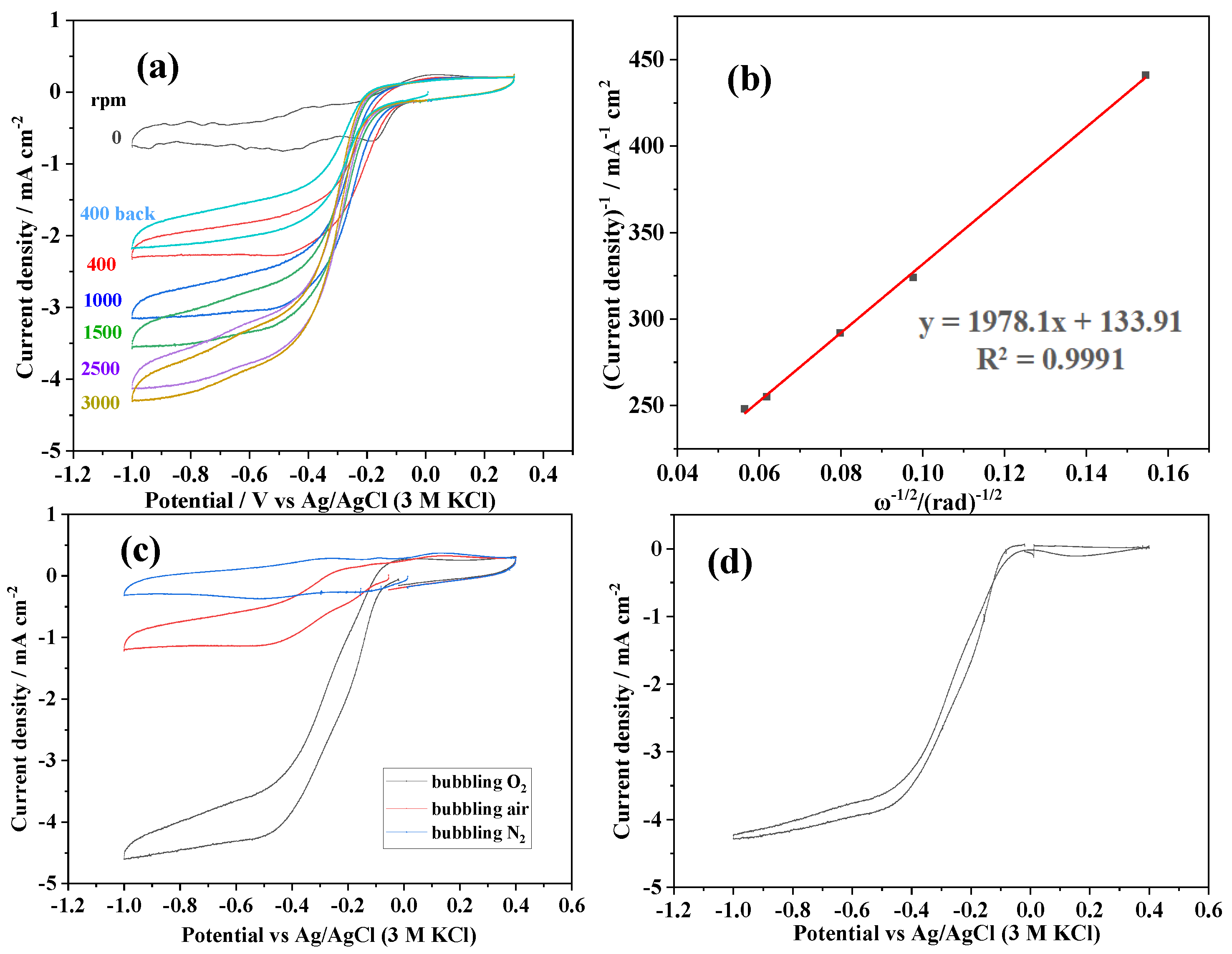
3.2.2. The Effect of Ink Drop Volume on RDE
3.2.3. The First Scan of CV Test
- -
- Without VXC-72 in the ink, the first and second cathodic peak seem to integrate into one broad peak. The onset potential is slightly more negative than for VXC-72 in the ink.
- -
- Without LSMO0.3 in the ink, there is only one sharp cathodic peak corresponding to the first cathodic peak in the case of LSMO0.3 in the ink. The onset potential is more negative, namely there is more overpotential, which also demonstrates that the VXC-72 alone has catalytic activity for oxygen reduction, though lower than that of LSMO0.3.
- -
- The situation is similar for bare glassy carbon which also has a weak oxygen reduction activity.
3.3. The Comparison of the Electroactivity of the LSMOx
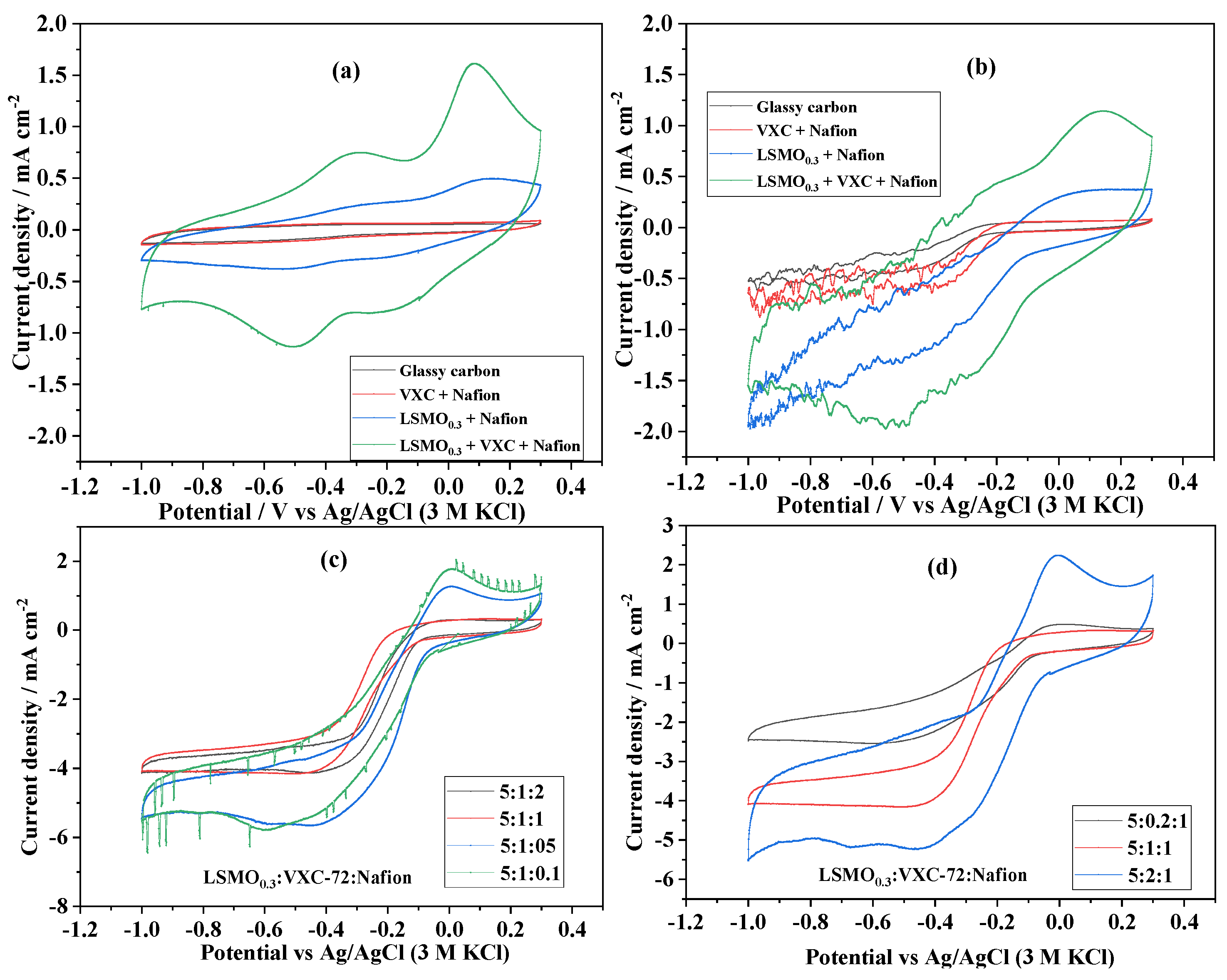
3.3.1. Effect of the Ink Composition and Formulation
- -
- 75 mg LSMO0.3 + 15 mg VXC-72 + 15 mg Nafion, i.e., the complete formulation
- -
- 75 mg LSMO0.3 + 15 mg Nafion, i.e., no carbon
- -
- 15 mg VXC-72 + 15 mg Nafion, i.e., no catalyst
3.3.2. Koutecký–Levich Analysis
3.3.3. Proposition of Net Electroactivity Comparison
4. Conclusions
Funding
Conflicts of Interest
Abbreviations
| ORR | oxygen reduction reaction |
| RDE | rotating disk electrode |
| LSV | linear sweep voltammetry |
| CV | cyclic voltammetry |
| LSMOx | La1−xSrxMnO3 |
References
- Dai, Y.; Yu, J.; Cheng, C.; Tan, P. Ni, Mini-review of perovskite oxides as oxygen electrocatalysts for rechargeable zinc–air batteries. Chem. Eng. J. 2020, 397, 125516. [Google Scholar] [CrossRef]
- Chu, W.; Higgins, D.; Chen, Z.; Cai, R. Non-precious Metal Oxides and Metal Carbides for ORR in Alkaline-Based Fuel Cells. In Non-Noble Metal Fuel Cell Catalysts; Chen, Z., Dodelet, J., Zhang, J., Eds.; Wiley: Hoboken, NJ, USA, 2014; pp. 357–388. [Google Scholar]
- Schmidt, T.J.; Gasteiger, H.A.; Stäb, G.D.; Urban, P.M.; Kolb, D.M.; Behm, R.J. Characterization of high-surface-area electrocatalysts using a rotating disk electrode configuration. J. Electrochem. Soc. 1998, 145, 2354. [Google Scholar] [CrossRef]
- Paulus, U.A.; Schmidt, T.J.; Gasteiger, H.A.; Behm, R.J. Oxygen reduction on a high-surface area Pt/Vulcan carbon catalyst: A thin-film rotating ring-disk electrode study. J. Electroanal. Chem. 2001, 495, 2134–2145. [Google Scholar] [CrossRef]
- Suntivich, J.; Gasteiger, H.; Yabuuchi, N.; Shao-Horn, Y. Electrocatalytic Measurement Methodology of Oxide Catalysts Using a Thin-Film Rotating Disk Electrode. J. Electrochem. Soc. 2010, 157, B1263–B1268. [Google Scholar] [CrossRef]
- Stoerzinger, K.; Lü, W.; Li, C.; Ariando; Venkatesan, T.; Shao-Horn, Y. Highly Active Epitaxial La(1–x)SrxMnO3 Surfaces for the Oxygen Reduction Reaction: Role of Charge Transfer. J. Phys. Chem. Lett. 2015, 6, 1435–1440. [Google Scholar] [CrossRef] [PubMed]
- Suntivich, J.; Gasteiger, H.; Yabuuchi, N.; Nakanishi, H.; Goodenough, J.; Shao-Horn, Y. Design principles for oxygen-reduction activity on perovskite oxide catalysts for fuel cells and metal–air batteries. Nat. Chem. 2011, 3, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, S.; Shimanoe, K.; Teraoka, Y.; Yamazoe, N. Oxygen Reduction Property of Ultrafine LaMnO3 Dispersed on Carbon Support. Electrochem. Solid-State Lett. 2005, 8, A270. [Google Scholar] [CrossRef]
- Kéranguéven, G.; Royer, S.; Savinova, E. Synthesis of efficient Vulcan–LaMnO3 perovskite nanocomposite for the oxygen reduction reaction. Electrochem. Commun. 2015, 50, 28–31. [Google Scholar] [CrossRef]
- Poux, T.; Napolskiy, F.; Dintzer, T.; Kéranguéven, G.; Istomin, S.; Tsirlina, G.; Antipov, E.; Savinova, E. Dual role of carbon in the catalytic layers of perovskite/carbon composites for the electrocatalytic oxygen reduction reaction. Catal. Today 2012, 189, 83–92. [Google Scholar] [CrossRef]
- Ge, X.; Du, Y.; Li, B.; Hor, T.; Sindoro, M.; Zong, Y.; Zhang, H.; Liu, Z. Intrinsically conductive perovskite oxides with enhanced stability and electrocatalytic activity for oxygen reduction reactions. ACS Catal. 2016, 6, 7865–7871. [Google Scholar] [CrossRef]
- Miyahara, Y.; Miyazaki, K.; Fukutsuka, T.; Abe, T. Influence of Surface Orientation on the Catalytic Activities of La0.8Sr0.2CoO3 Crystal Electrodes for Oxygen Reduction and Evolution Reactions. ChemElectroChem 2016, 3, 214–217. [Google Scholar] [CrossRef]
- Miyahara, Y.; Miyazaki, K.; Fukutsuka, T.; Abe, T. Catalytic roles of perovskite oxides in electrochemical oxygen reactions in alkaline media. J. Electrochem. Soc. 2014, 161, F694. [Google Scholar] [CrossRef]
- Elumeeva, K.; Masa, J.; Sierau, J.; Tietz, F.; Muhler, M.; Schuhmann, W. Perovskite-based bifunctional electrocatalysts for oxygen evolution and oxygen reduction in alkaline electrolytes. Electrochim. Acta 2016, 208, 25–32. [Google Scholar] [CrossRef]
- Athayde, D.; Souza, D.; Silva, A.; Vasconcelos, D.; Nunes, E.; da Costa, J.D.; Vasconcelos, W. Review of perovskite ceramic synthesis and membrane preparation methods. Ceram. Int. 2016, 42, 6555–6571. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Alonso, J.; Bian, J. Recent Advances in Perovskite-Type Oxides for Energy Conversion and Storage Applications. Adv. Energy Mater. 2021, 11, 2000459. [Google Scholar] [CrossRef]
- Xu, X.; Wang, W.; Zhou, W.; Shao, Z. Recent advances in novel nanostructuring methods of perovskite electrocatalysts for energy-related applications. Small Methods 2018, 2, 1800071. [Google Scholar] [CrossRef]
- Xu, X.; Su, C.; Shao, Z. Fundamental Understanding and Application of Ba(0.5)Sr(0.5)Co(0.8)Fe(0.2)O(3-δ) Perovskite in Energy Storage and Conversion: Past, Present, and Future. Energ. Fuel 2021, 35, 13585–13609. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, X.; Yao, S.; Hao, C.; Pan, C.; Xiang, X.; Tian, Z.; Shen, P.; Shao, Z.; Jiang, S. Boosting Electrocatalytic Activity of Single Atom Catalysts Supported on Nitrogen-Doped Carbon through N Coordination Environment Engineering. Small 2022, 18, 2105329. [Google Scholar] [CrossRef]
- Beall, C.E.; Fabbri, E.; Schmidt, T.J. Perovskite Oxide Based Electrodes for the Oxygen Reduction and Evolution Reactions: The Underlying Mechanism. ACS Catal. 2021, 11, 3094–3114. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhou, W.; Chen, Y.; Yu, J.; Xu, X.; Su, C.; Tadé, M.O.; Shao, Z. Boosting oxygen reduction reaction activity of palladium by stabilizing its unusual oxidation states in perovskite. Chem. Mater. 2015, 27, 3048–3054. [Google Scholar] [CrossRef]
- Xue, Y.; Sun, S.; Wang, Q.; Dong, Z.; Liu, Z. Transition metal oxide-based oxygen reduction reaction electrocatalysts for energy conversion systems with aqueous electrolytes. J. Mater. Chem. A 2018, 6, 10595–10626. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, P.; Dai, S. Recent Advances of Lanthanum-Based Perovskite Oxides for Catalysis. ACS Catal. 2015, 5, 6370–6385. [Google Scholar] [CrossRef]
- Pavone, M.; Muñoz-García, A.; Ritzmann, A.; Carter, E. First-Principles Study of Lanthanum Strontium Manganite: Insights into Electronic Structure and Oxygen Vacancy Formation. J. Phys. Chem. C 2014, 118, 13346–13356. [Google Scholar] [CrossRef]
- Tulloch, J.; Donne, S. Activity of perovskite La1−xSrxMnO3 catalysts towards oxygen reduction in alkaline electrolytes. J. Power Sources 2009, 188, 359–366. [Google Scholar] [CrossRef]
- Zhao, Y.; Hang, Y.; Zhang, Y.; Wang, Z.; Yao, Y.; He, X.; Zhang, C.; Zhang, D. Strontium-doped perovskite oxide La1−xSrxMnO3 (x = 0, 0.2, 0.6) as a highly efficient electrocatalyst for nonaqueous Li-O2 batteries. Electrochim. Acta 2017, 232, 296–302. [Google Scholar] [CrossRef]
- Xue, Y.; Miao, H.; Sun, S.; Wang, Q.; Li, S.; Liu, Z. (La1−xSrx)0.98MnO3 perovskite with A-site deficiencies toward oxygen reduction reaction in aluminum-air batteries. J. Power Sources 2017, 342, 192–201. [Google Scholar] [CrossRef]
- Deganello, F.; Marcì, G.; Deganello, G. Citrate–nitrate auto-combustion synthesis of perovskite-type nanopowders: A systematic approach. J. Eur. Ceram. Soc. 2009, 29, 439–450. [Google Scholar] [CrossRef]
- Garsany, Y.; Singer, I.L.; Swider-Lyons, K.E. Impact of film drying procedures on RDE characterization of Pt/VC electrocatalysts. J. Electroanal. Chem. 2011, 662, 396–406. [Google Scholar] [CrossRef]
- Xia, W.; Mahmood, A.; Liang, Z.; Zou, R.; Guo, S. Earth-Abundant Nanomaterials for Oxygen Reduction. Angew. Chem. Int. Ed. 2016, 55, 650–2676. [Google Scholar] [CrossRef]
- Brezesinski, T.; Wang, J.; Poiieux, J.; Dunn, B.; Tolbert, S. Templated Nanócrystal-Based Porous TiO2 Films for Next-Generation Electrochemical Capacitors. J. Am. Chem. Soc. 2009, 131, 1802–1809. [Google Scholar] [CrossRef]
- Zhang, Y.; Feng, F.; Zhang, C.; Zheng, Q.; Wang, C.; Hu, H.; Wu, M.; Guo, Y. Enhanced Catalytic Activity of LaMnO3 by A-Site Substitution as Air Electrode of Zn–Air Batteries with Attractive Durability. Energy Fuels 2020, 34, 10170–10177. [Google Scholar] [CrossRef]
- Luo, F.; Huang, Y.; Yan, C.; Jiang, S.; Li, X.; Wang, Z.; Liao, C. Molten alkali metal nitrate flux to well-crystallized and homogeneous La0. 7Sr0. 3MnO3 nanocrystallites. J. Magn. Magn. Mater. 2003, 260, 173–180. [Google Scholar] [CrossRef]
- Guo, J.; Jing, Y.; Shen, T.; Luo, H.; Liang, J.; Yuan, S. Effect of doped strontium on catalytic properties of La1–xSrxMnO3 for rhodamine B degradation. J. Rare Earth. 2021, 39, 1362–1369. [Google Scholar] [CrossRef]
- Jiang, S. Development of lanthanum strontium manganite perovskite cathode materials of solid oxide fuel cells: A review. J. Mater. Sci. 2008, 43, 6799–6833. [Google Scholar] [CrossRef]
- Chen, X.; Khor, K.; Chan, S. Electrochemical behavior of La(Sr)MnO3 electrode under cathodic and anodic polarization. Solid State Ionics 2004, 167, 379–387. [Google Scholar] [CrossRef]
- Chen, S.; Sheng, W.; Yabuuchi, N.; Ferreira, P.; Allard, L.; Shao-Horn, Y. Origin of Oxygen Reduction Reaction Activity on “Pt3Co” Nanoparticles: Atomically Resolved Chemical Compositions and Structures. J. Phys. Chem. C 2009, 113, 1109–1125. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Xu, T.; Wen, S.; Pan, M. Structure-dependent electrocatalytic activity of La1−xSrxMnO3 for oxygen reduction reaction. Sci China Chem. 2015, 58, 871–878. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | [25] | [26] | [27] | [38] | This Work |
|---|---|---|---|---|---|
| Catalysts | La0.6Sr0.4MnO3 | La0.4Sr0.6MnO3 | (La1− xSrx)0.98MnO3 | La1−xSrxMnO3 | La0.7Sr0.3MnO3 |
| Synthesis method | co-precipitation | Sol-gel method | solid–liquid method | modified polymer complex method | Citrate solution method |
| Catalyst loading | 21.3 ± 0.4 μg on 5 mm diameter GC electrode | 0.5 ± 0.1 mg cm−2 | 0.125 mg cm −2 | 0.5102 mg cm−2 | 0.893 mg cm−2 |
| Electrolyte | O2-saturated 1.0 M KOH | O2-saturated 0.1 M KOH | O2-saturated 0.1 M KOH | O2-staturated 1 M KOH | O2-staturated 1 M KOH |
| Scan rate | 5 mV s−1 | 10 mV s−1 | 5 mV s −1 | 5 mV s−1 | 10 mV s−1 |
| Rotation rate | 1500 rpm | 1600 rpm | 1600 rpm | 1600 rpm | 1500 rpm |
| Transferred electron number | close to 4 | 3.69 | 3.86 | close to 4 | 3.96 |
| Half-wave potential | - | - | 0.588 V vs. RHE | −0.127 V vs. Hg/HgO | −0.237 V vs. Ag/AgCl |
| Onset potential | - | −0.274 V vs. SCE | 0.903 V vs. RHE | 0.147 V vs. Hg/HgO | −0.096 V vs. Ag/AgCl |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, M. Case Study on the Reproducibility of Characterizing the Electrocatalytic Activity of La1−xSrxMnO3 in ORR. Crystals 2023, 13, 74. https://doi.org/10.3390/cryst13010074
Gong M. Case Study on the Reproducibility of Characterizing the Electrocatalytic Activity of La1−xSrxMnO3 in ORR. Crystals. 2023; 13(1):74. https://doi.org/10.3390/cryst13010074
Chicago/Turabian StyleGong, Minhui. 2023. "Case Study on the Reproducibility of Characterizing the Electrocatalytic Activity of La1−xSrxMnO3 in ORR" Crystals 13, no. 1: 74. https://doi.org/10.3390/cryst13010074
APA StyleGong, M. (2023). Case Study on the Reproducibility of Characterizing the Electrocatalytic Activity of La1−xSrxMnO3 in ORR. Crystals, 13(1), 74. https://doi.org/10.3390/cryst13010074