
Synthesis and Structural Analysis of Chiral Bis-dihydro[1,3]-naphthoxazines and Imidazolidine Derivatives Prepared by Three-Component Mannich-Type Condensation

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemistry (General Methods)
2.2. Synthesis of Bis-dihydro[1,3]naphthoxazines
2.2.1. Synthesis of 3,10-Bis((S)-1-phenylethyl)-2,3,4,9,10,11-hexa-hydronaphtho[1,2-e:4,3-e′]bis([1,3]oxazine) (3)
2.2.2. Synthesis of 3,9-Bis((S)-1-phenylethyl)-2,3,4,8,9,10-hexahydronaphtho[1,2-e:5,6-e′]bis([1,3]oxazine) (5)
2.3. Synthesis of Cyclohexane-1,2-diamine Derivatives
2.3.1. Synthesis of 1,1′-(((3aR,7aR)-Hexahydro-1H-benzo[d]imidazole-1,3(2H)-diyl)-bis(methylene))bis(naphthalen-2-ol) ((R,R)-9)
2.3.2. Synthesis of 1,1′-(((3aS,7aS)-Hexahydro-1H-benzo[d]imidazole-1,3(2H)-diyl)-bis(methylene))bis(naphthalen-2-ol) ((S,S)-9)
2.4. General Procedure for Enantioselective Addition of Diethylzinc to Aldehydes
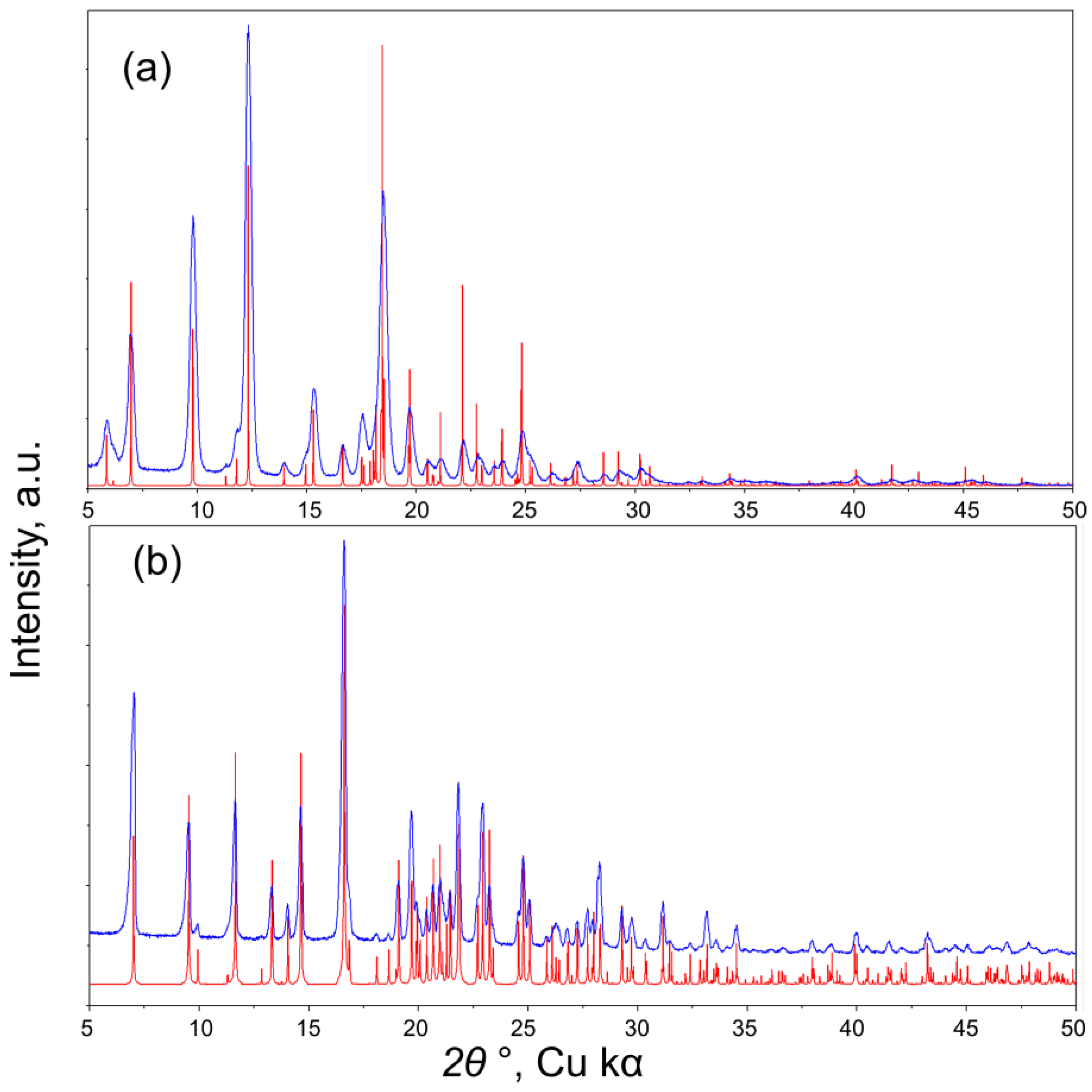
2.5. Powder X-ray Diffraction (PXRD)
2.6. Single-Crystal X-ray Diffraction (SCXRD)
2.7. Thermal Analysis (DSC)
3. Results and Discussion
3.1. Synthesis
3.2. Enantioselective Addition of Et2Zn to Aldehydes Catalyzed by Ligands (R,R)-9 and (S,S)-9
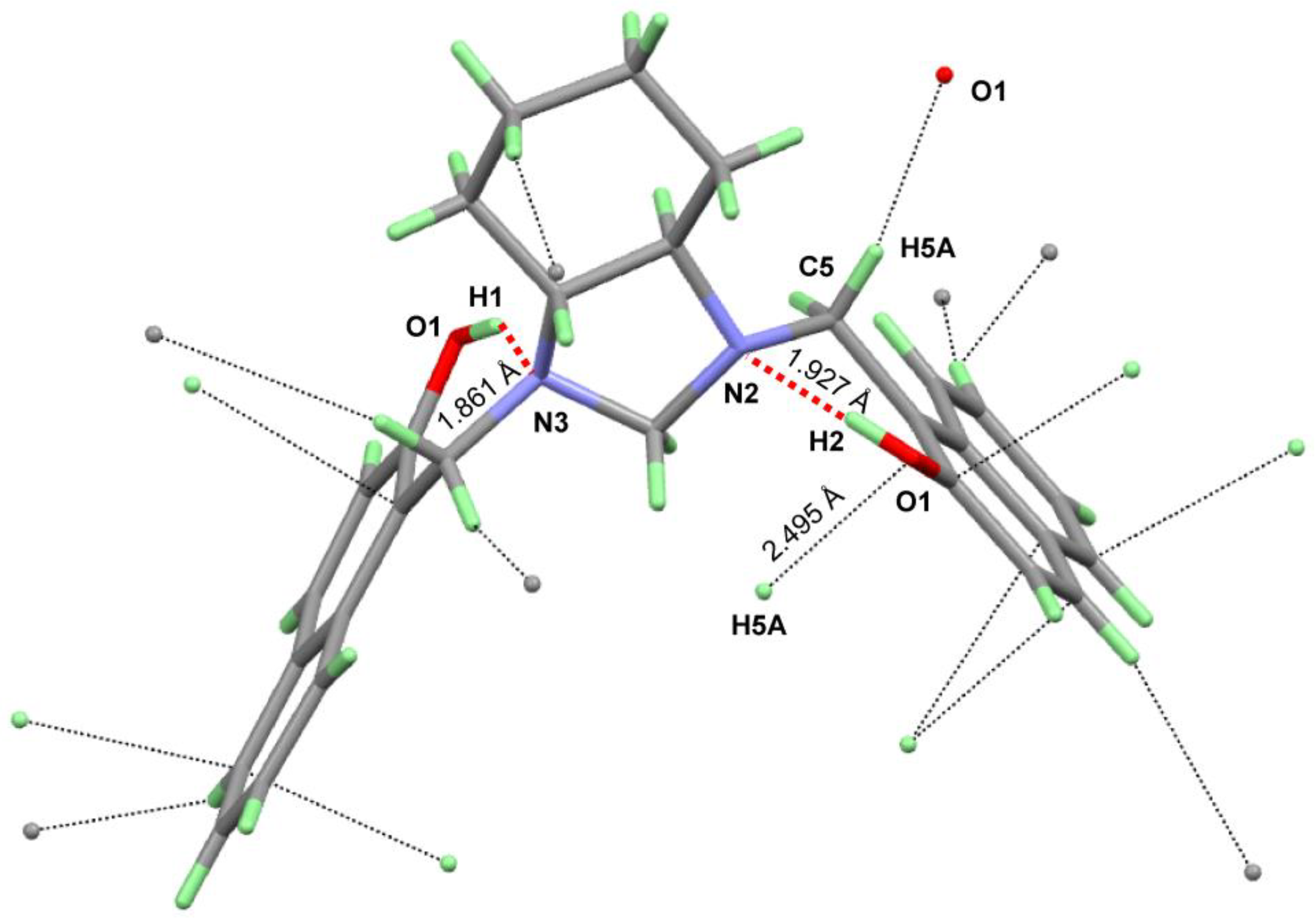
3.3. Crystal Structure Description
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rodrigues, M.O.; Eberlin, M.N.; Neto, B.A. How and why to investigate multicomponent reactions mechanisms? A critical review. Chem. Rec. 2021, 21, 2762–2781. [Google Scholar] [CrossRef] [PubMed]
- Insuasty, D.; Castillo, J.; Becerra, D.; Rojas, H.; Abonia, R. Synthesis of biologically active molecules through multicomponent reactions. Molecules 2020, 25, 505. [Google Scholar] [CrossRef] [PubMed]
- Lathwal, A.; Mathew, B.P.; Nath, M. Syntheses, biological and material significance of dihydro[1,3]oxazine derivatives: An overview. Curr. Org. Chem. 2021, 25, 133–174. [Google Scholar] [CrossRef]
- Shen, S.B.; Ishida, H. Synthesis and characterization of polyfunctional naphthoxazines and related polymers. J. Appl. Polym. Sci. 1996, 61, 1595–1605. [Google Scholar] [CrossRef]
- Liu, D.-X.; Zhang, L.-C.; Wang, Q.; Da, C.-S.; Xin, Z.-Q.; Wang, R.; Choi, M.C.; Chan, A.S. The application of chiral aminonaphthols in the enantioselective addition of diethylzinc to aryl aldehydes. Org. Lett. 2001, 3, 2733–2735. [Google Scholar] [CrossRef]
- Tavlinova-Kirilova, M.; Marinova, M.; Angelova, P.; Kamenova-Nacheva, M.; Kostova, K.; Dimitrov, V. Three component condensation of a Betti-type–efficient tool for synthesis of chiral naphthoxazines and aminobenzylnaphthols for enantioselective diethylzinc addition to aldehydes. Bulg. Chem. Commun. 2016, 48, 705–712. [Google Scholar]
- Marinova, M.; Kostova, K.; Tzvetkova, P.; Tavlinova-Kirilova, M.; Chimov, A.; Nikolova, R.; Shivachev, B.; Dimitrov, V. Synthesis of 1, 3-aminonaphthols by diastereoselective Betti-type aminoalkylation of dihydroxy naphthalenes; diastereoselectivity, absolute configuration, and application. Tetrahedron Asymmetry 2013, 24, 1453–1466. [Google Scholar] [CrossRef]
- Gualandi, A.; Calogero, F.; Potenti, S.; Cozzi, P.G. Al (salen) metal complexes in stereoselective catalysis. Molecules 2019, 24, 1716. [Google Scholar] [CrossRef]
- Bikas, R.; Emami, M.; Ślepokura, K.; Noshiranzadeh, N. Preparing Mn (III) salen-type Schiff base complexes using 1, 3-oxazines obtained by Mannich condensation: Towards removing ortho-hydroxyaldehydes. New J. Chem. 2017, 41, 9710–9717. [Google Scholar] [CrossRef]
- CrysAlis PRO; Rigaku Oxford Diffraction Ltd., UK Ltd.: Yarnton, UK, 2021.
- Bruker, S. APEX3 V2016. 9-0, SAINT V8. 37A; Bruker AXS Inc.: Madison, WI, USA, 2016; Volume 2013, p. 2014. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Farrugia, L. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Kitamura, M.; Suga, S.; Kawai, K.; Noyori, R. Catalytic asymmetric induction. Highly enantioselective addition of dialkylzincs to aldehydes. J. Am. Chem. Soc. 1986, 108, 6071–6072. [Google Scholar] [CrossRef]
- Noyori, R.; Kitamura, M. Enantioselective addition of organometallic reagents to carbonyl compounds: Chirality transfer, multiplication, and amplification. Angew. Chem. Int. Ed. Engl. 1991, 30, 49–69. [Google Scholar] [CrossRef]
- Genov, M.; Dimitrov, V.; Ivanova, V. New δ-aminoalcohol for the enantioselective addition of dialkylzincs to aldehydes. Tetrahedron Asymmetry 1997, 8, 3703–3706. [Google Scholar] [CrossRef]
- Ramachandran, G.; Sathiyanarayanan, K.I.; Sathishkumar, M.; Rathore, R.S.; Giridharan, P. Dual behavior of ammonium acetate for the synthesis of diverse symmetrical/unsymmetrical bis[1,3]oxazines possessing anticancer activity. Synth. Commun. 2015, 45, 2227–2239. [Google Scholar] [CrossRef]
- Ejfler, J.; Krauzy-Dziedzic, K.; Szafert, S.; Lis, T.; Sobota, P. Novel chiral and achiral benzoxazine monomers and their thermal polymerization. Macromolecules 2009, 42, 4008–4015. [Google Scholar] [CrossRef]
- Buckley, B.R.; Boxhall, J.Y.; Page, P.C.B.; Chan, Y.; Elsegood, M.R.; Heaney, H.; Holmes, K.E.; McIldowie, M.J.; McKee, V.; McGrath, M.J. Mannich and O-alkylation reactions of tetraalkoxyresorcin[4]arenes–the use of some products in ligand-assisted reactions. Eur. J. Org. Chem. 2006, 22, 5117–5134. [Google Scholar] [CrossRef]
- Barluenga, J.; Joglar, J.; González, F.J.; Fustero, S.; Krüger, C.; Tsay, Y.-H. Synthesis of 1, 3-amino alcohols from 2-aza-1, 3-dienes by reduction of 5, 6-dihydro-2H-1, 3-oxazines. Synthesis 1991, 387–393. [Google Scholar] [CrossRef]
- Hua, B.; Ding, Y.; Alimi, L.O.; Moosa, B.; Zhang, G.; Baslyman, W.S.; Sessler, J.; Khashab, N.M. Tuning the porosity of triangular supramolecular adsorbents for superior haloalkane isomer separations. Chem. Sci. 2021, 12, 12286–12291. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Alimi, L.O.; Du, J.; Hua, B.; Dey, A.; Yu, P.; Khashab, N.M. Pillar[3]trianglamines: Deeper cavity triangular macrocycles for selective hexene isomer separation. Chem. Sci. 2022, 13, 3244–3248. [Google Scholar] [CrossRef] [PubMed]
- Rivera, A.; Quiroga, D.; Ríos-Motta, J.; Kučeraková, M.; Dušek, M. meso-4, 4′-Dimethoxy-2, 2′-{[(3aR, 7aS)-2, 3, 3a, 4, 5, 6, 7, 7a-octahydro-1H-benzimidazole-1, 3-diyl] bis (methylene)} diphenol. Acta Crystallogr. E Crystallogr. Commun. 2013, 69, o1057–o1058. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Some Characteristic Protons | 8 | 9 |
|---|---|---|
| 1H NMR (Number of Protons) | 1H NMR (Number of Protons) | |
| C(H)-11 | 4.36 (4H) | 4.21 (2H) 4.39 (2H) |
| C(H)-13 | 3.01–3.16 (2H) | 2.45–2.60 (2H) |
| C(H)-16 | 4.98 (4H) | 3.66 (2H) |
| № | HCHO (Equivalent) | Solvent | Time (h) | T (°C) | Molar Ratio 8:9 3 | Yield 5 of 9 (%) |
|---|---|---|---|---|---|---|
| 1 | 10 1 | EtOH | 3 | 20 | 5:2 | - |
| 2 | 10 1 | EtOH | 5 | 20 | 2:1 | - |
| 3 | 10 1 | EtOH | 1 | 50 | 5:2 | - |
| 4 | 10 1 | EtOH | 6 | 50 | 2:1 | - |
| 5 | 10 1 | EtOH | 18 | 50 | 20:19 4 | - |
| 6 | 10 2 | EtOH | 24 | 50 | 7:10 | 34 6 |
| 7 | 10 1 | EtOH | 2.5 | reflux | 2:5 | - |
| 8 | 10 2 | toluene | 24 | 50 | 5:1 | - |
| 9 | 2 1 | MeOH | 2 | reflux | 0:1 | 50 7 |
| 10 | 2 2 | MeOH | 2 | reflux | 0:1 | 46 7 |
| 11 | 2 2 | EtOH | 2 | reflux | 0:1 | 34 7 |
| 12 | 2 2 | EtOH | 24 | 50 | 0:1 | 63 6 |
| Entry | Aldehyde | Ligands | Time (h) | Yield 11 (%) 1 | ee (%) Configuration |
|---|---|---|---|---|---|
| 1 | 2-Methoxybezaldehyde | (R,R)-9 | 72 | 66 | 53 (R) 2 |
| 2 | 2-Methoxybezaldehyde | (S,S)-9 | 24 | 80 | 58 (S) 2 |
| 3 | 1-Naphthaldehyde | (R,R)-9 | 52 | 45 | 39 (R) 3 |
| 4 | 1-Naphthaldehyde | (S,S)-9 | 120 | 54 | 30 (S) 3 |
| 5 | 4-Chlorobenzaldehyde | (R,R)-9 | 144 | 90 | 49 (R) 3 |
| 6 | 4-Chlorobenzaldehyde | (S,S)-9 | 120 | 30 | 50 (S) 3 |
| 7 | Cyclohexanecaboxaldehyde | (R,R)-9 | 24 | 46 | 38 (S) 2 |
| 8 | Cyclohexanecaboxaldehyde | (S,S)-9 | 24 | 49 | 40 (R) 2 |
| 9 | Ferrocenecarboxaldehyde | (R,R)-9 | 48 | 79 | 38 (R) 3 |
| 10 | Ferrocenecarboxaldehyde | (S,S)-9 | 120 | 74 | 42 (S) 3 |
| Compound 3 | Compound (R,R)-9 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bond Lengths, Å | Bond Lengths, Å | ||||||||||
| Atom | Atom | Å | Atom | Atom | Å | Atom | Atom | Å | Atom | Atom | Å |
| O1 | C3 | 1.373(5) | N22 | C23 | 1.466(5) | O1 | C25 | 1.360(4) | C1 | C5 | 1.510(3) |
| O2 | C2 | 1.363(5) | N12 | C13 | 1.491(6) | O2 | C26 | 1.357(3) | C7 | C8 | 1.511(3) |
| C3 | C2 | 1.420(6) | N22 | C24 | 1.490(6) | N2 | C5 | 1.468(3) | C9 | C10 | 1.422(4) |
| O1 | C21 | 1.456(6) | C3 | C4 | 1.362(6) | N3 | C7 | 1.475(3) | C3 | C4 | 1.419(4) |
| O2 | C32 | 1.456(6) | C2 | C1 | 1.367(6) | N2 | C6 | 1.476(3) | C15 | C16 | 1.508(3) |
| N12 | C32 | 1.429(6) | C7 | C8 | 1.381(9) | N3 | C6 | 1.468(3) | C19 | C20 | 1.523(5) |
| N12 | C11 | 1.467(6) | C24 | C25 | 1.528(7) | N3 | C15 | 1.467(3) | C17 | C18 | 1.527(4) |
| N22 | C21 | 1.431(5) | C13 | C14 | 1.521(6) | N2 | C16 | 1.471(3) | C18 | C19 | 1.524(5) |
| Bond Angles, ° | Bond Angles, ° | ||||||||||
| Atom | Atom | Atom | ° | Atom | Atom | Atom | ° | ||||
| C2 | O2 | C32 | 113.6(4) | C6 | N3 | C7 | 113.0(2) | ||||
| C3 | O1 | C21 | 114.0(3) | C6 | N2 | C5 | 113.1(2) | ||||
| N12 | C32 | O2 | 113.7(4) | N3 | C6 | N2 | 106.1(2) | ||||
| N12 | C11 | C1 | 111.9(4) | N3 | C15 | C20 | 117.3(2) | ||||
| C32 | N12 | C11 | 108.3(3) | N2 | C16 | C17 | 118.0(2) | ||||
| N12 | C13 | C15 | 109.0(4) | C25 | C8 | C7 | 121.6(3) | ||||
| N22 | C23 | C4 | 111.2(3) | C26 | C1 | C5 | 118.7(2) | ||||
| C21 | N22 | C23 | 107.9(3) | C16 | C17 | C18 | 107.6(3) | ||||
| N22 | C24 | C26 | 110.3(4) | C15 | C20 | C19 | 108.8(3) | ||||
| D | H | A | d(D-H)/Å | d(H-A)/Å | d(D-A)/Å | D-H-A/° |
|---|---|---|---|---|---|---|
| O1 | H1 | N3 | 0.82 | 1.86 | 2.596(3) | 148.4 |
| O2 | H2 | N2 | 0.82 | 1.93 | 2.650(3) | 146.4 |
| C5 | H5A | O2 1 | 0.97 | 2.49 | 3.223(3) | 131.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tavlinova-Kirilova, M.; Dikova, K.; Marinova, M.K.; Kamenova-Nacheva, M.; Rusew, R.; Sbirkova-Dimitrova, H.; Shivachev, B.; Kostova, K.; Dimitrov, V. Synthesis and Structural Analysis of Chiral Bis-dihydro[1,3]-naphthoxazines and Imidazolidine Derivatives Prepared by Three-Component Mannich-Type Condensation. Crystals 2023, 13, 1495. https://doi.org/10.3390/cryst13101495
Tavlinova-Kirilova M, Dikova K, Marinova MK, Kamenova-Nacheva M, Rusew R, Sbirkova-Dimitrova H, Shivachev B, Kostova K, Dimitrov V. Synthesis and Structural Analysis of Chiral Bis-dihydro[1,3]-naphthoxazines and Imidazolidine Derivatives Prepared by Three-Component Mannich-Type Condensation. Crystals. 2023; 13(10):1495. https://doi.org/10.3390/cryst13101495
Chicago/Turabian StyleTavlinova-Kirilova, Maya, Krasimira Dikova, Maya K. Marinova, Mariana Kamenova-Nacheva, Rusi Rusew, Hristina Sbirkova-Dimitrova, Boris Shivachev, Kalina Kostova, and Vladimir Dimitrov. 2023. "Synthesis and Structural Analysis of Chiral Bis-dihydro[1,3]-naphthoxazines and Imidazolidine Derivatives Prepared by Three-Component Mannich-Type Condensation" Crystals 13, no. 10: 1495. https://doi.org/10.3390/cryst13101495
APA StyleTavlinova-Kirilova, M., Dikova, K., Marinova, M. K., Kamenova-Nacheva, M., Rusew, R., Sbirkova-Dimitrova, H., Shivachev, B., Kostova, K., & Dimitrov, V. (2023). Synthesis and Structural Analysis of Chiral Bis-dihydro[1,3]-naphthoxazines and Imidazolidine Derivatives Prepared by Three-Component Mannich-Type Condensation. Crystals, 13(10), 1495. https://doi.org/10.3390/cryst13101495