
Two Crystal Forms of 4′-Methyl-2,4-dinitrodiphenylamine: Polymorphism Governed by Conformational Flexibility of a Supramolecular Synthon


Abstract
:1. Introduction
2. Materials and Methods
2.1. Crystallization of Polymorphic Forms
2.2. Single-Crystal X-ray Crystallography
2.3. Computational Details
3. Results and Discussion
3.1. Molecular Geometry
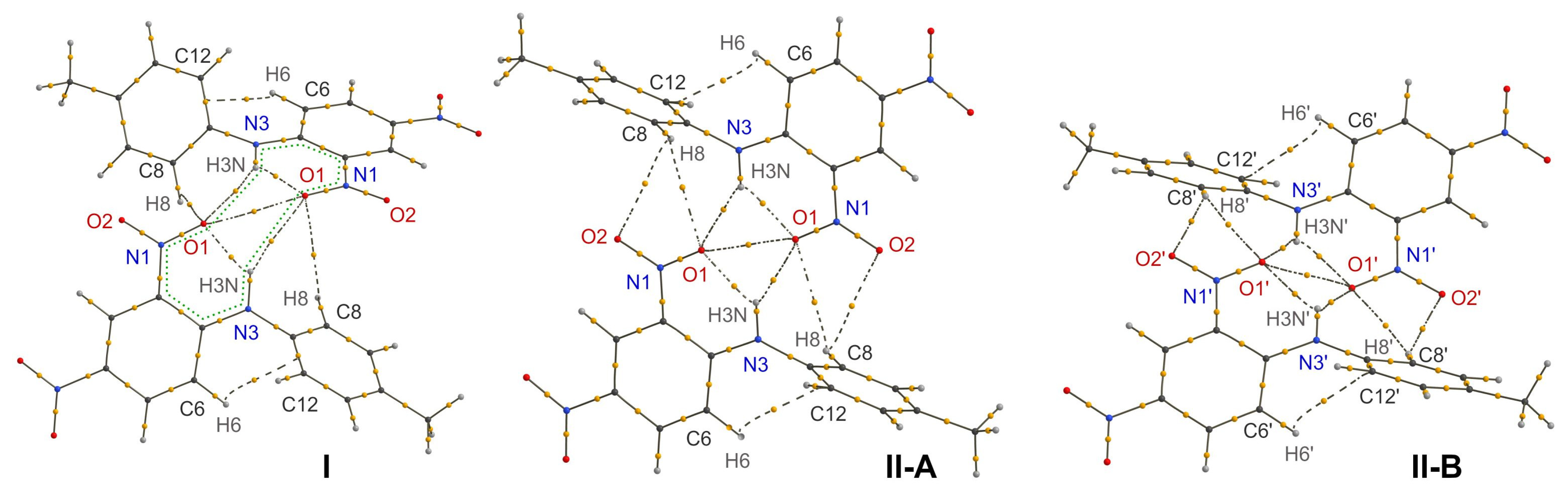
3.2. Intermolecular H-Bonds
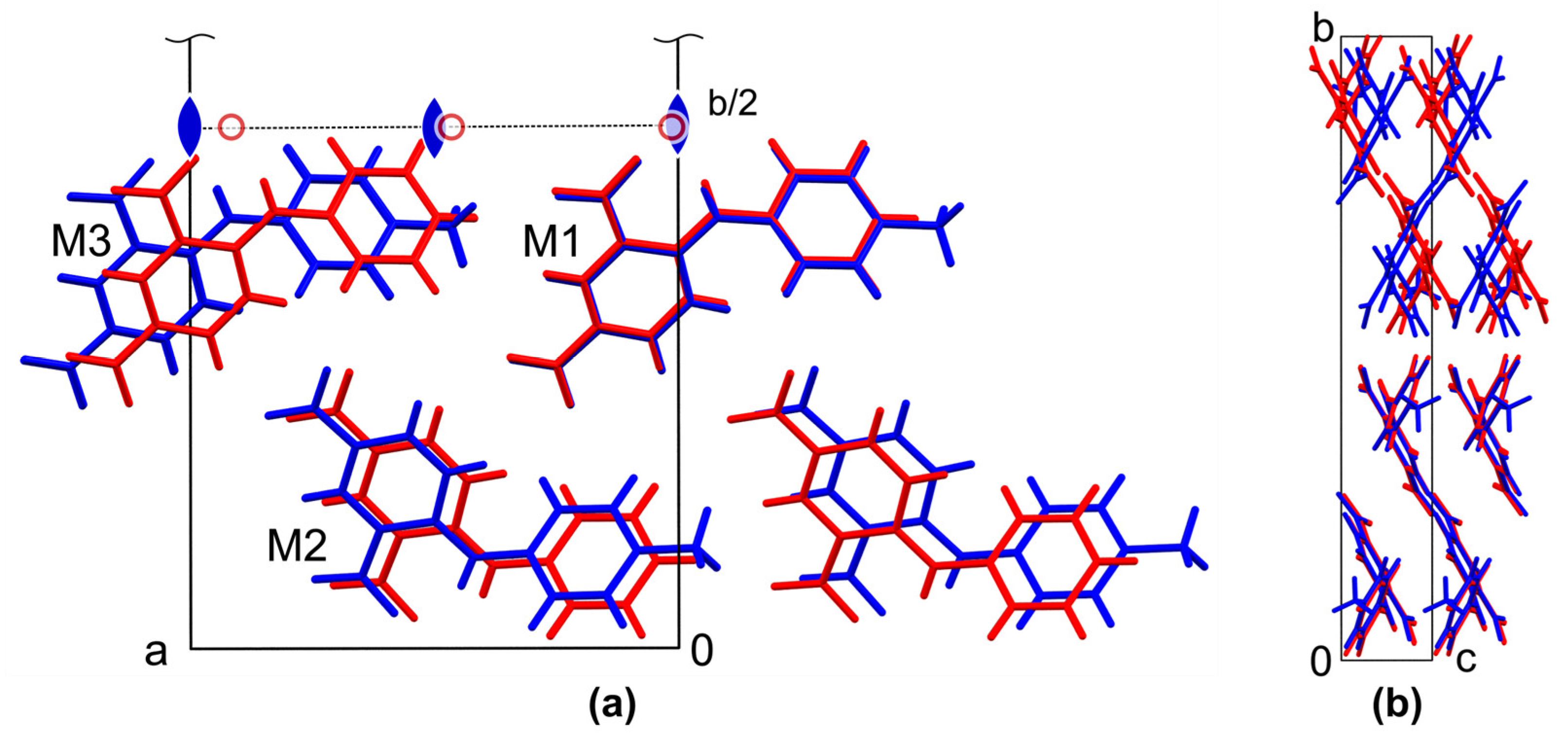
3.3. Other Intermolecular Interactions and Crystal Packing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bernstein, J. Polymorphism in Molecular Crystals; International Union of Crystallography Monographs on Crystallography; Oxford University Press: Oxford, UK; Oxford Clarendon Press: New York, NY, USA, 2002; ISBN 0-19-850605-8. [Google Scholar]
- Bernstein, J. Crystal Growth, Polymorphism and Structure-Property Relationships in Organic Crystals. J. Phys. D Appl. Phys. 1993, 26, B66–B76. [Google Scholar] [CrossRef]
- Hilfiker, R. (Ed.) Polymorphism in the Pharmaceutical Industry; Wiley-VCH: Weinheim, Germany, 2006; ISBN 978-3-527-31146-0. [Google Scholar]
- Politzer, P.; Murray, J.S. Energetic Materials. Pt. 1, Decomposition, Crystal and Molecular Properties; Elsevier: Amsterdam, The Netherlands; Heidelberg, Germany, 2003; ISBN 0-444-51518-6. [Google Scholar]
- Ok, K.M.; Chi, E.O.; Halasyamani, P.S. Bulk Characterization Methods for Non-Centrosymmetric Materials: Second-Harmonic Generation, Piezoelectricity, Pyroelectricity, and Ferroelectricity. Chem. Soc. Rev. 2006, 35, 710. [Google Scholar] [CrossRef] [PubMed]
- McCrone, W.C. Polymorphism. In Physics and Chemistry of the Organic Solid State; Fox, D., Labes, M.M., Weissberger, A., Eds.; Interscience Publishers: London, UK, 1965; Volume 2, pp. 725–767. [Google Scholar]
- Price, S.L. Why Don’t We Find More Polymorphs? Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2013, 69, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Cabeza, A.J.; Bernstein, J. Conformational Polymorphism. Chem. Rev. 2014, 114, 2170–2191. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R. Supramolecular Synthons in Crystal Engineering—A New Organic Synthesis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Jetti, R.K.R.; Boese, R.; Sarma, J.A.R.P.; Reddy, L.S.; Vishweshwar, P.; Desiraju, G.R. Searching for a Polymorph: Second Crystal Form of 6-Amino-2-Phenylsulfonylimino-1,2-Dihydropyridine. Angew. Chem. Int. Ed. 2003, 42, 1963–1967. [Google Scholar] [CrossRef]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Gilday, L.C.; Robinson, S.W.; Barendt, T.A.; Langton, M.J.; Mullaney, B.R.; Beer, P.D. Halogen Bonding in Supramolecular Chemistry. Chem. Rev. 2015, 115, 7118–7195. [Google Scholar] [CrossRef]
- Rijhwani, J.B.; Asthana, J.G. Synthesis and Antibacterial Activity of Some 2,4-Dinitrodiphenylamine Derivatives. Indian Drugs Pharm. Ind. 1978, 13, 41–42. [Google Scholar]
- Manea-Saghin, A.; Andreea Marin, C.; Pădurețu, C.; Kajzar, F. Third-Order Optical Nonlinearity of DNA-CTMA Complex Doped with Different Aromatic Moieties. Opt. Laser Technol. 2022, 149, 107863. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, Z.; Zhong, X.; Huang, L.; Xue, W.; Li, M. Application of Powder Test to the Study of New Organic Materials for Frequency Doubling. Chin. J. Lasers Chin. 1992, 19, 627–633. [Google Scholar]
- Vizgert, R.V.; Davydov, B.L.; Kotovshchikov, S.G.; Starodubtseva, M.P. Generation of the Second Harmonic of a Neodymium Laser in Powders of Noncentrosymmetric Organic Compounds. Sov. J. Quantum Electron. 1982, 12, 214–215. [Google Scholar] [CrossRef]
- Hernández-Paredes, J.; Carrillo-Torres, R.C.; Hernández-Negrete, O.; Sotelo-Mundo, R.R.; Glossman-Mitnik, D.; Esparza-Ponce, H.E.; Alvarez-Ramos, M.E. Experimental and Theoretical Study on the Molecular Structure, Covalent and Non-Covalent Interactions of 2,4-Dinitrodiphenylamine: X-Ray Diffraction and QTAIM Approach. J. Mol. Struct. 2017, 1141, 53–63. [Google Scholar] [CrossRef]
- Ikramuddeen, T.M.; Chandrasekara, N.; Ramarajan, K.; Subramanian, K.A. Kinetics of Nucleophilic Substitution Reaction of 1-Chloro-2,4-Dinitrobenzene with Substituted Anilines. J. Indian Chem. Soc. 1989, 66, 342–344. [Google Scholar]
- Bruker. SAINT; Bruker AXS Inc.: Madison, WI, USA, 2013. [Google Scholar]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of Silver and Molybdenum Microfocus X-Ray Sources for Single-Crystal Structure Determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-Mechanical Condensed Matter Simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Peintinger, M.F.; Oliveira, D.V.; Bredow, T. Consistent Gaussian Basis Sets of Triple-Zeta Valence with Polarization Quality for Solid-State Calculations. J. Comput. Chem. 2013, 34, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Differences of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules—A Quantum Theory; Oxford University Press: Oxford, UK, 1990; ISBN 0-19-855168-1. [Google Scholar]
- Gatti, C.; Saunders, V.R.; Roetti, C. Crystal Field Effects on the Topological Properties of the Electron Density in Molecular Crystals: The Case of Urea. J. Chem. Phys. 1994, 101, 10686. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll, version 19.10.12; TK Gristmill Software: Overland Park, KS, USA, 2019. [Google Scholar]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen Bond Strengths Revealed by Topological Analyses of Experimentally Observed Electron Densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Spackman, M.A. How Reliable Are Intermolecular Interaction Energies Estimated from Topological Analysis of Experimental Electron Densities? Cryst. Growth Des. 2015, 15, 5624–5628. [Google Scholar] [CrossRef]
- Ananyev, I.V.; Karnoukhova, V.A.; Dmitrienko, A.O.; Lyssenko, K.A. Toward a Rigorous Definition of a Strength of Any Interaction Between Bader’s Atomic Basins. J. Phys. Chem. A 2017, 121, 4517–4522. [Google Scholar] [CrossRef]
- Lyssenko, K.A. Analysis of Supramolecular Architectures: Beyond Molecular Packing Diagrams. Mendeleev Commun. 2012, 22, 1–7. [Google Scholar] [CrossRef]
- Bruno, I.J.; Cole, J.C.; Kessler, M.; Luo, J.; Motherwell, W.D.S.; Purkis, L.H.; Smith, B.R.; Taylor, R.; Cooper, R.I.; Harris, S.E.; et al. Retrieval of Crystallographically-Derived Molecular Geometry Information. J. Chem. Inf. Comput. Sci. 2004, 44, 2133–2144. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and Decoding Hydrogen-Bond Patterns of Organic Compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Fedyanin, I.V.; Lyssenko, K.A. New Hydrogen-Bond-Aided Supramolecular Synthon: A Case Study of 2,4,6-Trinitroaniline. CrystEngComm 2013, 15, 10086–10093. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Fedyanin, I.V.; Karnoukhova, V.A.; Lyssenko, K.A. Conformational Analysis of a Supramolecular Synthon: A Case Study of 8-Hydroxyquinoline. CrystEngComm 2018, 20, 652–660. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Vologzhanina, A.V. Intermolecular Interactions in Functional Crystalline Materials: From Data to Knowledge. Crystals 2019, 9, 478. [Google Scholar] [CrossRef]
- Bombicz, P. The Way from Isostructurality to Polymorphism. Where Are the Borders? The Role of Supramolecular Interactions and Crystal Symmetries. Crystallogr. Rev. 2017, 23, 118–151. [Google Scholar] [CrossRef]
- Fedyanin, I.V. Control of Supramolecular Chirality in Co-Crystals of Achiral Molecules via Stacking Interactions and Hydrogen Bonding. CrystEngComm 2022, 24, 2591–2601. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2 | |
| Form I | Form II | ||
| Chemical formula | C13H11N3O4 | ||
| M | 273.25 | ||
| CCDC number/ CSD identifier | 1434709 | 1434708 | 1491537/ QQQFGG02 |
| Temperature, K | 100 | 100 | 100 |
| Crystal shape and color | red needle | orange prism | red needle |
| Crystal size, mm | 0.24 × 0.07 × 0.05 | 0.12 × 0.10 × 0.08 | |
| Crystal system | orthorhombic | triclinic | monoclinic |
| Space group | P21212 | P¯1 | P21/n |
| Z/Z’ | 4/1 | 4/2 | 4/1 |
| a, Å | 12.214(2) | 7.1979(4) | 3.7626(2) |
| b, Å | 26.118(5) | 12.8243(8) | 11.0374(5) |
| c, Å | 3.8006(7) | 13.1514(8) | 26.8216(12) |
| α, ° | 90 | 86.2870(10) | |
| β, ° | 90 | 83.6270(10) | 90.273(2) |
| γ, ° | 90 | 86.2960(10) | |
| V, Å3 | 1212.4(4) | 1201.93(12) | 1113.87(9) |
| dcalc, g cm−3 | 1.497 | 1.510 | |
| μ (MoKα), mm−1 | 0.114 | 0.115 | |
| 2θ max (°) | 60 | 60 | |
| Measured reflections | 8306 | 25991 | |
| Independent reflections | 3550 | 6939 | |
| R(int) | 0.077 | 0.033 | |
| No. of parameters | 186 | 371 | |
| Reflections with I > 2σ(I) | 2070 | 5500 | |
| R1 | 0.055 | 0.053 | |
| wR2 | 0.094 | 0.136 | |
| GOF | 0.99 | 1.03 | |
| ρmax/ρmin (e Ǻ−3) | 0.29/−0.30 | 0.44/−0.26 | |
| I | II-A | II-B | ||||
|---|---|---|---|---|---|---|
| exp. | DFT | exp. | DFT | exp. | DFT | |
| Intramolecular H-bond | ||||||
| N···O, Å | 2.620(3) | 2.593 | 2.6308(19) | 2.606 | 2.6295(19) | 2.614 |
| H···O, Å | 1.813 | 1.802 | 1.867 | 1.828 | 1.877 | 1.842 |
| N−H···O, ° | 133.87 | 132.04 | 129.48 | 130.80 | 128.42 | 130.21 |
| ρ(r), e Å−3 | 0.266 | 0.252 | 0.244 | |||
| ∇2ρ(r), e Å−3 | 3.42 | 3.29 | 3.21 | |||
| ε | 0.02 | 0.02 | 0.04 | |||
| EEML, kcal/mol | 12.27 | 11.36 | 10.85 | |||
| Intermolecular H-bond | ||||||
| N···O, Å | 3.330(3) | 3.457 | 3.2558(19) | 3.300 | 3.3865(19) | 3.436 |
| H···O, Å | 2.425 | 2.564 | 2.328 | 2.389 | 2.543 | 2.600 |
| N−H···O, ° | 148.22 | 146.75 | 151.58 | 149.03 | 140.33 | 139.61 |
| ρ(r), e Å−3 | 0.034 | 0.050 | 0.035 | |||
| ∇2ρ(r), e Å−3 | 0.54 | 0.83 | 0.53 | |||
| ε | 1.86 | 0.50 | 1.39 | |||
| EEML, kcal/mol | 0.86 | 1.31 | 0.86 | |||
| Intermolecular O1···O1 contact | ||||||
| O1···O1, Å | 2.699(3) | 2.891 | 2.683(2) | 2.744 | 2.928(2) | 2.923 |
| ρ(r), e Å−3 | 0.044 | 0.062 | 0.046 | |||
| ∇2ρ(r), e Å−3 | 0.87 | 1.25 | 0.85 | |||
| ε | 0.47 | 0.53 | 0.55 | |||
| EEML, kcal/mol | 1.45 | 2.33 | 1.43 | |||
| Intermolecular C8-H8···O1 contact | ||||||
| C···O, Å | 3.504(4) | 3.593 | 3.449(2) | 3.429 | 3.541(2) | 3.629 |
| H···O, Å | 2.886 | 3.048 | 2.849 | 2.792 | 2.623 | 2.720 |
| C-H···O, ° | 115.97 | 111.76 | 114.64 | 117.43 | 141.43 | 113.74 |
| ρ(r), e Å−3 | 0.022 | 0.037 | 0.037 | |||
| ∇2ρ(r), e Å−3 | 0.32 | 0.50 | 0.49 | |||
| ε | 0.30 | 0.14 | 0.78 | |||
| EEML, kcal/mol | 0.47 | 0.87 | 0.87 | |||
| Intermolecular C8-H8···O2 contact | ||||||
| C···O, Å | 3.659(4) | 3.775 | 3.390(2) | 3.458 | 3.431(2) | 3.510 |
| H···O, Å | 3.220 | 3.418 | 2.778 | 2.877 | 2.421 | 2.561 |
| C-H···O, ° | 105.15 | 100.84 | 115.34 | 141.28 | 145.04 | 145.66 |
| ρ(r), e Å−3 | 0.031 | 0.049 | ||||
| ∇2ρ(r), e Å−3 | 0.44 | 0.64 | ||||
| ε | 0.17 | 0.05 | ||||
| EEML, kcal/mol | 0.73 | 1.17 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fedyanin, I.V.; Samigullina, A.I. Two Crystal Forms of 4′-Methyl-2,4-dinitrodiphenylamine: Polymorphism Governed by Conformational Flexibility of a Supramolecular Synthon. Crystals 2023, 13, 296. https://doi.org/10.3390/cryst13020296
Fedyanin IV, Samigullina AI. Two Crystal Forms of 4′-Methyl-2,4-dinitrodiphenylamine: Polymorphism Governed by Conformational Flexibility of a Supramolecular Synthon. Crystals. 2023; 13(2):296. https://doi.org/10.3390/cryst13020296
Chicago/Turabian StyleFedyanin, Ivan V., and Aida I. Samigullina. 2023. "Two Crystal Forms of 4′-Methyl-2,4-dinitrodiphenylamine: Polymorphism Governed by Conformational Flexibility of a Supramolecular Synthon" Crystals 13, no. 2: 296. https://doi.org/10.3390/cryst13020296
APA StyleFedyanin, I. V., & Samigullina, A. I. (2023). Two Crystal Forms of 4′-Methyl-2,4-dinitrodiphenylamine: Polymorphism Governed by Conformational Flexibility of a Supramolecular Synthon. Crystals, 13(2), 296. https://doi.org/10.3390/cryst13020296