

Structural Characterization of Pharmaceutical Cocrystals with the Use of Laboratory X-ray Powder Diffraction Patterns

Abstract
:1. Introduction
2. Crystal Structures from Powder Diffraction—How Reliable Are They?
- (1)
- It should provide minimal discrepancies between the experimental and calculated patterns, i.e., minimal values of χ2 and profile R-factors, Rp and Rwp;
- (2)
- It must be in complete agreement with all experimental data on the other physicochemical studies;
- (3)
- The geometry of all molecular fragments, i.e., bond lengths and angles, and geometry of intermolecular contacts (H-bonds, π…π, dipole–dipole, and others weak interactions) must correspond to the known data, for example, collected in the Cambridge Structural Database (CSD [38]).
- (4)
- For the reliable crystal structure determined from XRPD data, the upper RMSCD limit should not exceed the value of 0.35 A.
- -
- When working with XRPD and a new polycrystalline sample, the search for its “correct” crystal structure is useless because nobody knows what it should look like. Instead, the search for the best structural model(s) suitable for Criteria (1)–(4) allows you to obtain a certain result;
- -
- Any additional information limiting the area of possible structural models has to be used;
- -
- The best structural model that has been obtained in several independent diffraction experiments with different samples of the substance, and which allows us to predict the properties of the bulk material, can be considered a reliable crystal structure.
3. Applications of SDPD Methods to Pharmaceutical Cocrystals and Salts
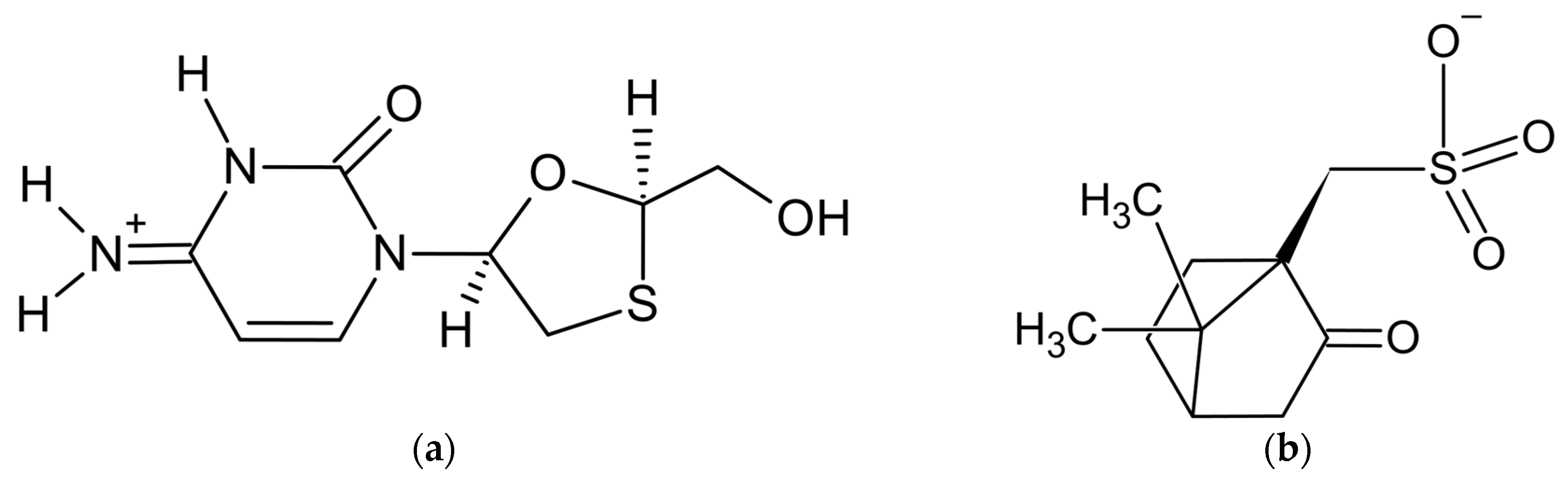
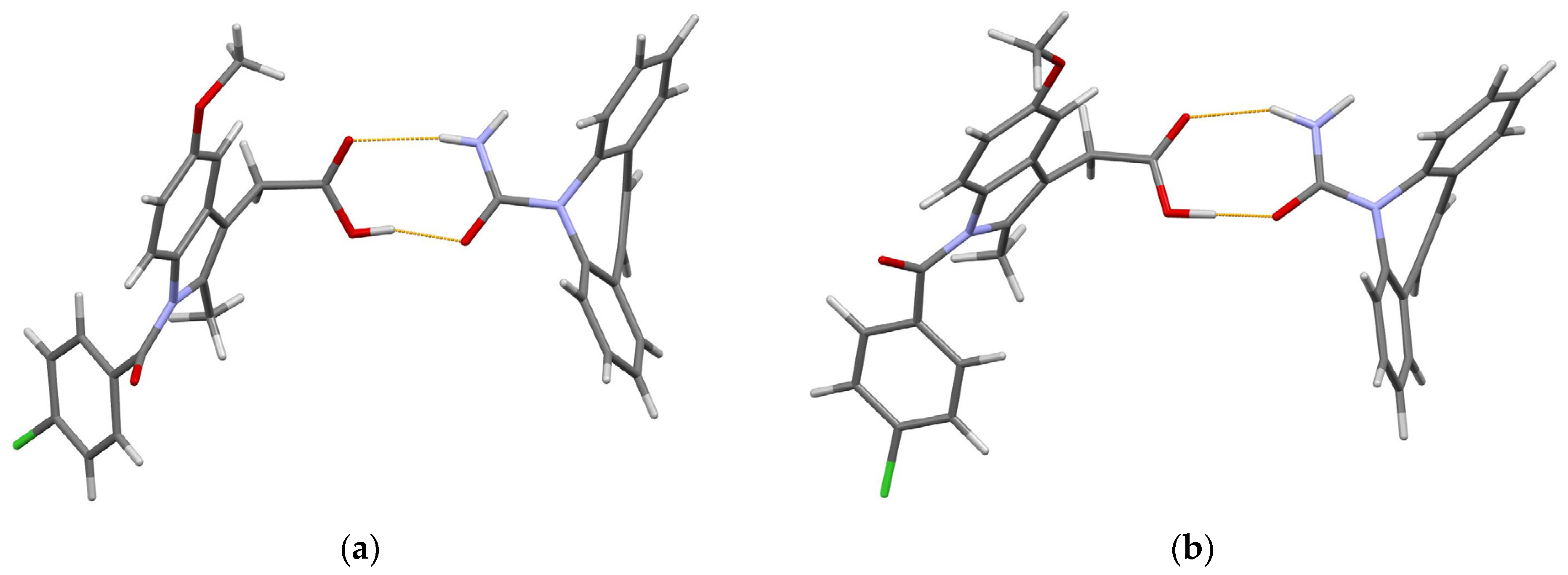
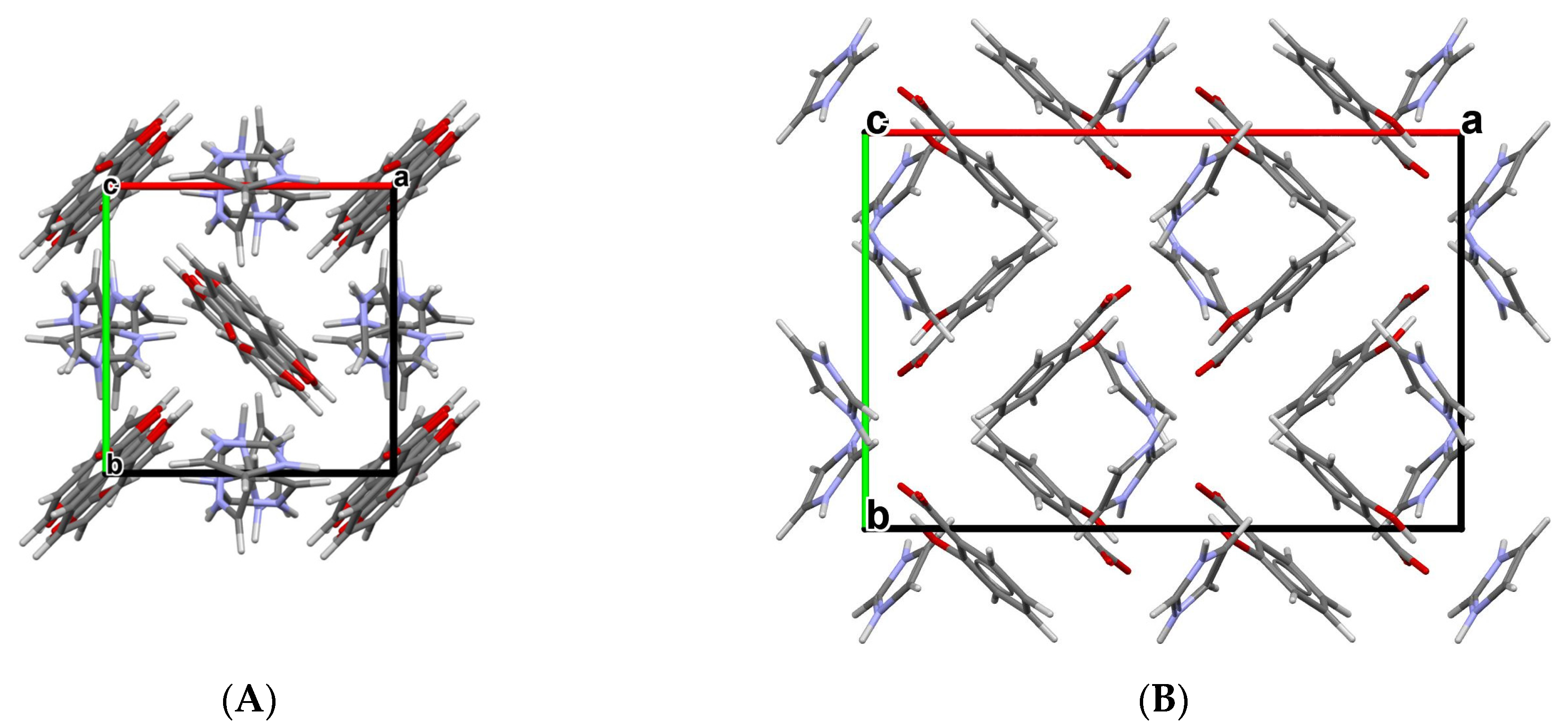
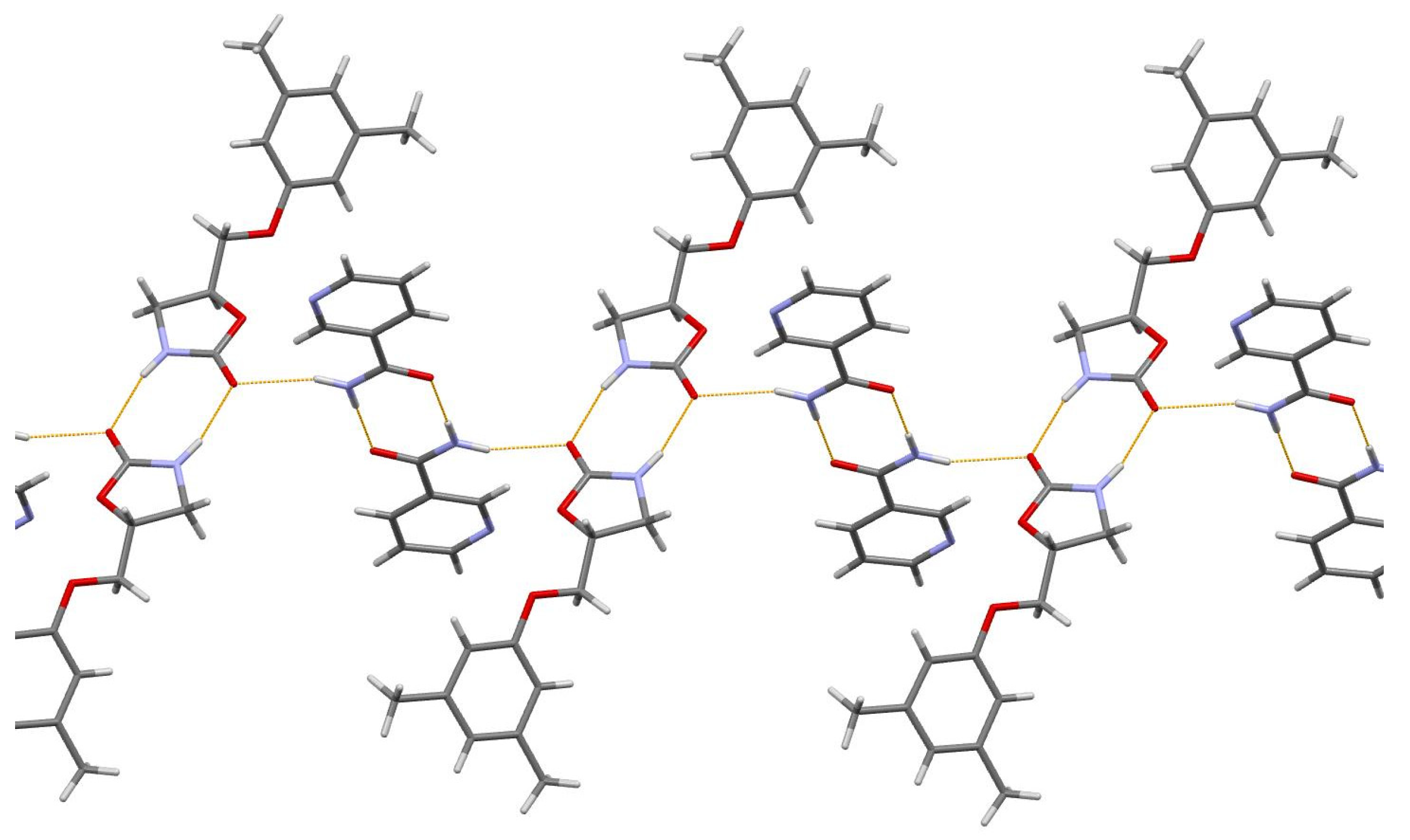
3.1. Lamivudine Camphorsulfonate (1:1) Salt
3.2. Furosemide Urea (1:1) and Carbamazepine Indomethacin (1:1)
3.3. Three Imidazole-Based (1:1) Salts of Salicylic Acid
3.4. Metaxalone Nicotinamide (1:1)
3.5. Acridine Diclofenac (1:1) Salt
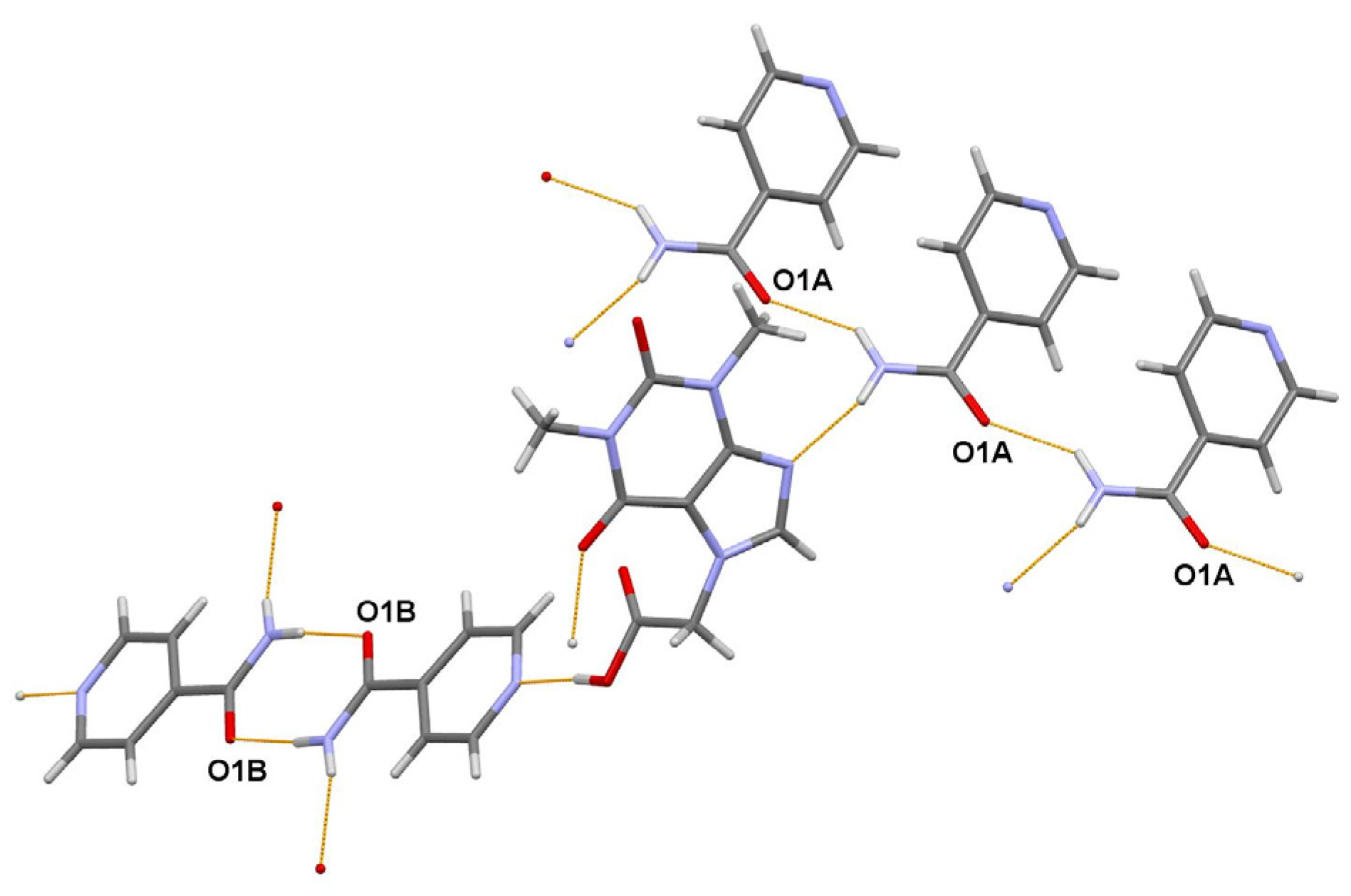
3.6. Acefylline Nicotinamide (1:2) Cocrystal
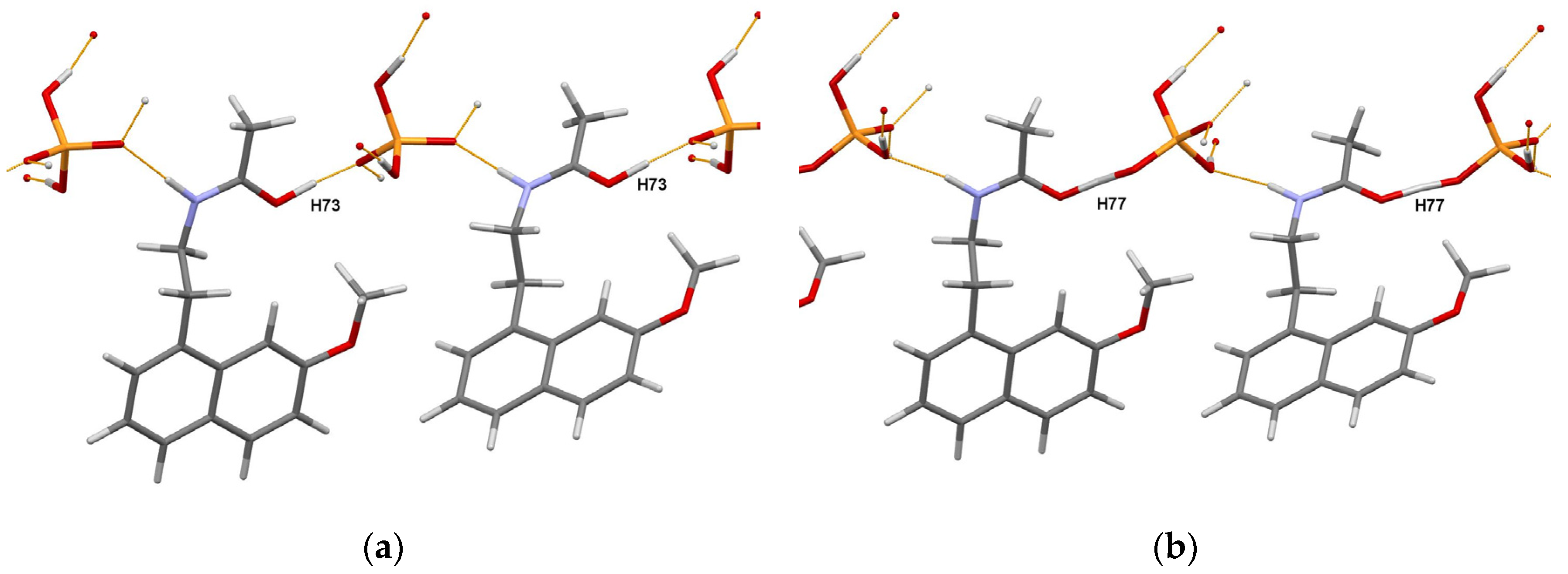
3.7. Salt–Cocrystal Continuum of the Agomelatine: Phosphoric Acid (1:1) System
4. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brittain, H.G.; Bogdanowich, S.J.; Bugay, D.E.; DeVincentis, J.; Lewen, G.; Newman, A.W. Physical characterization of pharmaceutical solids. Pharm. Res. 1991, 8, 963–973. [Google Scholar] [CrossRef]
- Brittain, H.G. X-Ray diffraction of pharmaceutical materials. Profiles Drug Subst. Excip. Relat. Methodol. 2003, 30, 271–319. [Google Scholar]
- Randall, C.S.; Rocco, W.L.; Ricou, P. XRD in pharmaceutical analysis: A versatile tool for problem-solving. Am. Pharm. Rev. 2010, 13, 52–59. [Google Scholar]
- Thakral, N.K.; Zanon, R.L.; Kelly, R.C.; Thakral, S. Applications of powder X-ray diffraction in small molecule pharmaceuticals: Achievements and aspirations. J. Pharm. Sci. 2018, 107, 2969–2982. [Google Scholar] [CrossRef]
- Rodríguez, I.; Gautam, R.; Tinoco, A.D. Using X-ray diffraction techniques for biomimetic drug development, formulation, and polymorphic characterization. Biomimetics 2021, 6, 1. [Google Scholar] [CrossRef]
- Thakral, S.; Terban, M.W.; Thakral, N.K.; Suryanarayanan, R. Recent advances in the characterization of amorphous pharmaceuticals by X-Ray diffractometry. Adv. Drug Deliv. Rev. 2016, 100, 183–193. [Google Scholar] [CrossRef]
- Datta, S.; Grant, D.J.W. Crystal structures of drugs: Advances in determination, prediction and engineering. Nat. Rev. Drug Disc. 2004, 3, 42–57. [Google Scholar] [CrossRef]
- Harris, K.D.M.; Tremayne, M. Crystal structure determination from powder diffraction data. Chem. Mater. 1996, 8, 2554–2570. [Google Scholar] [CrossRef]
- Harris, K.D.M.; Tremayne, M.; Kariuki, B.M. Contemporary advances in the use of powder X-ray diffraction for structure determination. Angew. Chem. Int. Ed. 2001, 40, 1626–1651. [Google Scholar] [CrossRef]
- Chernyshev, V.V. Structure determination from powder diffraction. Russ. Chem. Bull. 2001, 50, 2273–2292. [Google Scholar] [CrossRef]
- Harris, K.D.M. Modern applications of powder X-ray diffraction in pharmaceutical sciences. Am. Pharm. Rev. 2004, 7, 86–91. [Google Scholar]
- David, W.I.F.; Shankland, K.; McCusker, L.B.; Baerlocher, C. (Eds.) Structure Determination from Powder Diffraction Data; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Cerný, R.; Favré-Nicolin, V. Direct space methods of structure determination from powder diffraction: Principles, guidelines and perspectives. Z. Krist.-Cryst. Mater. 2007, 222, 105–113. [Google Scholar] [CrossRef]
- Tsue, H.; Horiguchi, M.; Tamura, R.; Fujii, K.; Uekusa, H. Crystal structure solution of organic compounds from X-ray powder diffraction data. J. Synth. Org. Chem. Jpn. 2007, 65, 1203–1212. [Google Scholar] [CrossRef]
- David, W.I.F.; Shankland, K. Structure determination from powder diffraction data. Acta Crystallogr. Sect. A 2008, 64, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Dinnebier, R.E.; Billinge, S.J.L. (Eds.) Powder Diffraction: Theory and Practice; RSC Publishing: Cambridge, UK, 2008. [Google Scholar]
- Cerný, R. Crystal structures from powder diffraction: Principles, difficulties and progress. Crystals 2017, 7, 142. [Google Scholar] [CrossRef] [Green Version]
- Altomare, A.; Ciriaco, F.; Cuocci, C.; Falcicchio, A.; Fanelli, F. Combined powder X-ray diffraction data and quantum-chemical calculations in EXPO2014. Powder Diffr. 2017, 32, S123–S128. [Google Scholar] [CrossRef]
- Kabova, E.A.; Cole, J.C.; Korb, O.; López-Ibáñez, M.; Williams, A.C.; Shankland, K. Improved performance of crystal structure solution from powder diffraction data through parameter tuning of a simulated annealing algorithm. J. Appl. Crystallogr. 2017, 50, 1411–1420. [Google Scholar] [CrossRef]
- Kabova, E.A.; Blundell, C.D.; Shankland, K. Pushing the limits of molecular crystal structure determination from powder diffraction data in high-throughput chemical environments. J. Pharm. Sci. 2018, 107, 2042–2047. [Google Scholar] [CrossRef] [Green Version]
- Haleblian, J.; McCrone, W. Pharmaceutical applications of polymorphism. J. Pharm. Sci. 1969, 58, 911–929. [Google Scholar] [CrossRef]
- Ainurofiq, A.; Dinda, K.E.; Pangestika, M.W.; Himawati, U.; Wardhani, W.D.; Sipahutar, Y.T. The effect of polymorphism on active pharmaceutical ingredients: A review. Int. J. Res. Pharm. Sci. 2020, 11, 1621–1630. [Google Scholar] [CrossRef]
- Park, H.; Kim, J.-S.; Hong, S.; Ha, E.-S.; Nie, H.; Zhou, Q.T.; Kim, M.-S. Tableting process-induced solid-state polymorphic transition. J. Pharm. Investig. 2022, 52, 175–194. [Google Scholar] [CrossRef]
- Braga, D.; Casali, L.; Grepioni, F. The relevance of crystal forms in the pharmaceutical field: Sword of Damocles or innovation tools? Int. J. Mol. Sci. 2022, 23, 9013. [Google Scholar] [CrossRef]
- Yao, C.; Zhang, S.; Wang, L.; Tao, X. Recent advances in polymorph discovery methods of organic crystals. Cryst. Growth Des. 2023, 23, 637–654. [Google Scholar] [CrossRef]
- Almarsson, Ö.; Zaworotko, M.J. Crystal engineering of the composition of pharmaceutical phases. Do pharmaceutical co-crystals represent a new path to improved medicines? Chem. Commun. 2004, 1889–1896. [Google Scholar] [CrossRef]
- Vishweshwar, P.; McMahon, J.A.; Bis, J.A.; Zaworotko, M.J. Pharmaceutical co-crystals. J. Pharm. Sci. 2006, 95, 499–516. [Google Scholar] [CrossRef]
- Brittain, H.G. Pharmaceutical cocrystals: The coming wave of new drug substances. J. Pharm. Sci. 2013, 102, 311–317. [Google Scholar] [CrossRef]
- Bolla, G.; Nangia, A. Pharmaceutical cocrystals: Walking the talk. Chem. Commun. 2016, 52, 8342–8360. [Google Scholar] [CrossRef]
- Kumar, S.; Nanda, A. Pharmaceutical cocrystals: An overview. Indian J. Pharm. Sci. 2017, 79, 858–871. [Google Scholar] [CrossRef]
- Vioglio, P.C.; Chierotti, M.R.; Gobetto, R. Pharmaceutical aspects of salt and cocrystal forms of APIs and characterization challenges. Adv. Drug Deliv. Rev. 2017, 117, 86–110. [Google Scholar] [CrossRef]
- Duggirala, N.K.; LaCasse, S.M.; Zaworotko, M.J.; Krzyzaniak, J.F.; Arora, K.K. Pharmaceutical cocrystals: Formulation approaches to develop robust drug products. Cryst. Growth Des. 2020, 20, 617–626. [Google Scholar] [CrossRef]
- Guo, M.; Sun, X.; Chen, J.; Cai, T. Pharmaceutical cocrystals: A review of preparations, physicochemical properties and applications. Acta Pharm. Sin. B 2021, 11, 2537–2564. [Google Scholar] [CrossRef]
- Teoh, Y.; Ayoub, G.; Huskić, I.; Titi, H.M.; Nickels, C.W.; Herrmann, B.; Friščić, T. SpeedMixing: Rapid tribochemical synthesis and discovery of pharmaceutical cocrystals without milling or grinding media. Angew. Chem. Int. Ed. 2022, 61, e202206293. [Google Scholar] [CrossRef]
- Bolla, G.; Sarma, B.; Nangia, A. Crystal engineering of pharmaceutical cocrystals in the discovery and development of improved drugs. Chem. Rev. 2022, 122, 11514–11603. [Google Scholar] [CrossRef]
- Munjal, B.; Suryanarayanan, R. Applications of synchrotron powder X-ray diffractometry in drug substance and drug product characterization. Trends Anal. Chem. 2021, 136, 116181. [Google Scholar] [CrossRef]
- Available online: https://www.imca.aps.anl.gov/IMCA-CAT (accessed on 20 February 2023).
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Van de Streek, J.; Neumann, M.A. Validation of molecular crystal structures from powder diffraction data with dispersion-corrected density functional theory (DFT-D). Acta Crystallogr. Sect. B 2014, 70, 1020–1032. [Google Scholar] [CrossRef]
- Reilly, A.M.; Cooper, R.I.; Adjiman, C.S.; Bhattacharya, S.; Boese, A.D.; Brandenburg, J.G.; Bygrave, P.J.; Bylsma, R.; Campbell, J.E.; Car, R.; et al. Report on the sixth blind test of organic crystal structure prediction methods. Acta Crystallogr. Sect. B 2016, 72, 439–459. [Google Scholar] [CrossRef]
- Pawley, G.S. Unit-cell refinement from powder diffraction scans. J. Appl. Crystallogr. 1981, 14, 357–361. [Google Scholar] [CrossRef]
- Le Bail, A.; Duroy, H.; Fourquet, J.L. Ab-initio structure determination of LiSbWO6 by X-ray powder diffraction. Mater. Res. Bull. 1988, 23, 447–452. [Google Scholar] [CrossRef]
- Habermehl, S.; Schlesinger, C.; Schmidt, M.U. Structure determination from unindexed powder data from scratch by a global optimization approach using pattern comparison based on cross-correlation functions. Acta Crystallogr. Sect. B 2022, B78, 195–213. [Google Scholar] [CrossRef]
- Schlesinger, C.; Fitterer, A.; Buchsbaum, C.; Habermehl, S.; Chierotti, M.R.; Nervi, C.; Schmidt, M.U. Ambiguous structure determination from powder data: Four different structural models of 4,11-difluoroquinacridone with similar X-ray powder patterns, fit to the PDF, SSNMR and DFT-D. IUCrJ 2022, 9, 406–424. [Google Scholar] [CrossRef]
- Altomare, A. Solving molecular compounds from powder diffraction data: Are results always reliable? IUCrJ 2022, 9, 403–405. [Google Scholar] [CrossRef]
- De Wolff, P.M. A simplified criterion for the reliability of a powder pattern indexing. J. Appl. Crystallogr. 1968, 1, 108–113. [Google Scholar] [CrossRef]
- Chan, F.C.; Anwar, J.; Cernik, R.; Barnes, P.; Wilson, R.M. Ab initio structure determination of sulfathiazole polymorph V from synchrotron X-ray powder diffraction data. J. Appl. Crystallogr. 1999, 32, 436–441. [Google Scholar] [CrossRef]
- Hughes, D.S.; Hursthouse, M.B.; Threlfall, T.; Tavener, S. A new polymorph of sulfathiazole. Acta Crystallogr. Sect. C 1999, 55, 1831–1833. [Google Scholar] [CrossRef]
- Llinas, A.; Box, K.J.; Burley, J.C.; Glen, R.; Goodman, J.M. A new method for the reproducible generation of polymorphs: Two forms of sulindac with very different solubilities. J. Appl. Crystallogr. 2007, 40, 379–381. [Google Scholar] [CrossRef]
- Grzesiak, A.L.; Matzger, A.J. New form discovery for the analgesis flurbiprofen and sulindac facilitated by polymer-induced heteronucleation. J. Pharm. Sci. 2007, 96, 2978–2986. [Google Scholar] [CrossRef] [Green Version]
- Chernyshev, V.V.; Machula, A.A.; Kukushkin, S.Y.; Velikodny, Y.A. Carvedilol dihydrogen phosphate hemihydrate: A powder study. Acta Crystallogr. Sect. E 2009, 65, o2020–o2021. [Google Scholar] [CrossRef] [Green Version]
- Vogt, F.G.; Copley, R.C.B.; Mueller, R.L.; Spoors, G.P.; Cacchio, T.N.; Carlton, R.A.; Katrincic, L.M.; Kennady, J.M.; Parsons, S.; Chetina, O.V. Isomorphism, disorder, and hydration in the crystal structures of racemic and single-enantiomer carvedilol phosphate. Cryst. Growth Des. 2010, 10, 2713–2733. [Google Scholar] [CrossRef]
- Bruning, J.; Alig, E.; Schmidt, M.U. Ezetimibe anhydrate, determined from laboratory powder diffraction data. Acta Crystallogr. Sect. C 2010, 66, o341–o344. [Google Scholar] [CrossRef]
- Shimpi, M.R.; Childs, S.L.; Bostrom, D.; Velaga, S.P. New cocrystals of ezetimibe with L-proline and imidazole. CrystEngComm 2014, 16, 8984–8993. [Google Scholar] [CrossRef]
- Bortolotti, M.; Lonardelli, I.; Pepponi, G. Determination of the crystal structure of nifedipine form C by synchrotron powder diffraction. Acta Crystallogr. Sect. B 2011, 67, 357–364. [Google Scholar] [CrossRef]
- Gunn, E.; Guzei, I.A.; Cai, T.; Yu, L. Polymorphism of nifedipine: Crystal structure and reversible transition of the metastable β polymorph. Cryst. Growth Des. 2012, 12, 2037–2043. [Google Scholar] [CrossRef]
- Prohens, R.; Barbas, R.; Portell, A.; Font-Bardia, M.; Alcobe, X.; Puigjaner, C. Polymorphism of cocrystals: The promiscuoud behavior of Agomelatine. Cryst. Growth Des. 2016, 16, 1063–1070. [Google Scholar] [CrossRef]
- Lee, M.-J.; Aitipamula, S.; Choi, G.J.; Chow, P.S. Agomelatine-hydroquinone (1:1) cocrystal: Novel polymorphs and their thermodynamic relationship. Acta Crystallogr. Sect. B 2019, 75, 969–977. [Google Scholar] [CrossRef] [Green Version]
- Schlesinger, C.; Bolte, M.; Schmidt, M.U. Challenging structure determination from powder diffraction data: Two pharmaceutical salts and one cocrystal with Z′ = 2. Z. Krist.-Cryst. Mater. 2019, 234, 257–268. [Google Scholar] [CrossRef]
- Boultif, A.; Louer, D. Indexing of powder diffraction patterns for low-symmetry lattices by the successive dichotomy method. J. Appl. Crystallogr. 1991, 24, 987–993. [Google Scholar] [CrossRef]
- David, W.I.F.; Shankland, K.; van de Streek, J.; Pidcock, E.; Motherwell, W.D.S.; Cole, J.C. DASH: A program for crystal structure determination from powder diffraction data. J. Appl. Crystallogr. 2006, 39, 910–915. [Google Scholar] [CrossRef]
- Coelho, A.A. TOPAS and TOPAS-Academic: An optimization program integrating computer algebra and crystallographic objects written in C++. J. Appl. Crystallogr. 2018, 51, 210–218. [Google Scholar] [CrossRef] [Green Version]
- Rahal, O.A.; Majumder, M.; Spillman, M.J.; van de Streek, J.; Shankland, K. Co-crystal structures of furosemide: Urea and carbamazepine:indomethacin determined from powder X-ray diffraction data. Crystals 2020, 10, 42. [Google Scholar] [CrossRef] [Green Version]
- Majumder, M.; Buckton, G.; Rawlinson-Malone, C.; Williams, A.C.; Spillman, M.J.; Shankland, N.; Shankland, K. A carbamazepine: Indomethacin (1:1) cocrystal produced by milling. CrystEngComm 2011, 13, 6327–6328. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Martins, I.C.B.; Al-Sabbagh, D.; Meyer, K.; Maiwald, M.; Scholz, G.; Emmerling, F. Insight into the structure and properties of novel imidazole-based salts of salicylic acid. Molecules 2019, 24, 4144. [Google Scholar] [CrossRef] [Green Version]
- Nabulsi, N.A.R.; Gandour, R.D.; Fronczek, F.R. CSD Communication (Private Communication). 2013. [Google Scholar]
- Martins, I.C.B.; Emmerling, F. Carbamazepine dihydroxybenzoic acid cocrystals: Exploring packing interactions and reaction kinetics. Cryst. Growth Des. 2021, 21, 6961–6970. [Google Scholar] [CrossRef]
- Gohel, S.K.; Palanisamy, V.; Sanphui, P.; Prakash, M.; Singh, G.P.; Chernyshev, V. Isostructural cocrystals of metaxalone with improved dissolution characteristics. RSC Adv. 2021, 11, 30689–30700. [Google Scholar] [CrossRef]
- Visser, J.W. A fully automatic program for finding the unit cell from powder data. J. Appl. Crystallogr. 1969, 2, 89–95. [Google Scholar] [CrossRef]
- Zhukov, S.G.; Babaev, E.V.; Chernyshev, V.V.; Rybakov, V.B.; Sonneveld, E.J.; Schenk, H. Crystal structure determination of 2-oxo-3-benzoyloxazolo[3,2-a]pyridine from X-ray powder data. Z. Krist.-Cryst. Mater. 2000, 215, 306–308. [Google Scholar] [CrossRef]
- Zlokazov, V.B.; Chernyshev, V.V. MRIA—A program for a full profile analysis of powder multiphase neutron-diffraction time-of-flight (direct and Fourier) spectra. J. Appl. Crystallogr. 1992, 25, 447–451. [Google Scholar] [CrossRef] [Green Version]
- Varsa, S.R.B.; Sanphui, P.; Chernyshev, V. Polymorphs and isostructural cocrystals of dexamethasone: Towards the improvement of aqueous solubility. CrystEngComm 2022, 24, 6045–6058. [Google Scholar] [CrossRef]
- Fayaz, T.K.S.; Palanisamy, V.; Sanphui, P.; Chernyshev, V. Multicomponent solid forms of antibiotic cephalexin towards improved chemical stability. CrystEngComm 2023, 25, 1252–1262. [Google Scholar] [CrossRef]
- Mirocki, A.; Conterosito, E.; Palin, L.; Sikorski, A.; Milanesio, M.; Lopresti, M. Crystal structure of a new 1:1 acridine-diclofenac salt, obtained with high yield by a mechanochemical approach. Crystals 2022, 12, 1573. [Google Scholar] [CrossRef]
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.; Rizzi, R.; Corriero, N.; Falcicchio, A. EXPO2013: A kit of tools for phasing crystal structures from powder data. J. Appl. Crystallogr. 2013, 46, 1231–1235. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef] [Green Version]
- Mirocki, A.; Lopresti, M.; Palin, L.; Conterosito, E.; Sikorski, A.; Milanesio, M. Exploring the molecular landscape of multicomponent crystals formed by naproxen drug and acridines. CrystEngComm 2022, 24, 6839–6853. [Google Scholar] [CrossRef]
- Allu, S.; Garai, A.; Chernyshev, V.V.; Nangia, A.K. Synthesis of ternary cocrystals, salts, and hydrates of acefylline with enhanced dissolution and high permeability. Cryst. Growth Des. 2022, 22, 4165–4181. [Google Scholar] [CrossRef]
- Werner, P.-E.; Eriksson, L.; Westdahl, M. TREOR, a semi-exhaustive trial-and-error powder indexing program for all symmetries. J. Appl. Crystallogr. 1985, 18, 367–370. [Google Scholar] [CrossRef]
- Voguri, R.S.; Ranga, S.; Dey, A.; Ghosal, S. Solid-state phase transition of agomelatine-phosphoric acid molecular complexes along the salt-cocrystal continuum: Ab initio powder X-ray diffraction structure determination and DFT-D2 analysis. Cryst. Growth Des. 2020, 20, 7647–7657. [Google Scholar] [CrossRef]
- Neumann, M.A. X-Cell: A novel indexing algorithm for routine tasks and difficult cases. J. Appl. Crystallogr. 2003, 36, 356–365. [Google Scholar] [CrossRef] [Green Version]
- MedeA, Materials Design, Inc.: San Diego, CA, USA, 2015.
- Stevens, J.S.; Byard, S.J.; Schroeder, S.L.M. Salt or co-crystal? Determination of protonation state by X-ray photoelectron spectroscopy (XPS). J. Pharm. Sci. 2010, 99, 4453–4457. [Google Scholar] [CrossRef]
- Harris, K.D.M. NMR Crystallography as a vital tool in assisting crystal structure determination from powder XRD data. Crystals 2022, 12, 1277. [Google Scholar] [CrossRef]
- Smalley, C.J.H.; Logsdail, A.J.; Hughes, C.E.; Iuga, D.; Young, M.T.; Harris, K.D.M. Solid-state structural properties of alloxazine determined from powder XRD data in conjunction with DFT-D calculations and solid-state NMR spectroscopy: Unraveling the tautomeric identity and pathways for tautomeric interconversion. Cryst. Growth Des. 2022, 22, 524–534. [Google Scholar] [CrossRef]
- Heng, T.; Yang, D.; Wang, R.; Zhang, L.; Lu, Y.; Du, G. Progress in research on Artificial Intelligence applied to polymorphism and cocrystal prediction. ACS Omega 2021, 6, 15543–15550. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substance | Powder Sample CCDC Ref. Code, Radiation, Temperature, Year of Publication | Single-Crystal Sample CCDC Ref. Code, Temperature, Year of Publication |
|---|---|---|
| Sulfathiazole, polymorph V | SUTHAZ06, synchrotron, RT, 1999 [47] | SUTHAZ05, T = 150 K, 1999 [48] |
| Sulindac, monoclinic polymorph | DOHREX01, laboratory X-ray, RT, 2007 [49] | DOHREX03, T = 123 K, 2007 [50] |
| Carvedilol dihydrogen phosphate hemihydrate | XOZJOM, laboratory X-ray, RT, 2009 [51] | XOZJOM01, T = 90 K, 2010 [52] |
| Ezetimibe | QUWYIR, laboratory X-ray, RT, 2010 [53] | QUWYIR01, RT, 2014 [54] |
| Nifedipine, triclinic polymorph | BICCIZ01, synchrotron, RT, 2011 [55] | BICCIZ03, RT, 2012 [56] |
| Agomelatine–hydroquinone (1:1) cocrystal, polymorph II | FAFGOL01, synchrotron, T = 100 K, 2016 [57] | FAFGOL02, RT, 2019 [58] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chernyshev, V.V. Structural Characterization of Pharmaceutical Cocrystals with the Use of Laboratory X-ray Powder Diffraction Patterns. Crystals 2023, 13, 640. https://doi.org/10.3390/cryst13040640
Chernyshev VV. Structural Characterization of Pharmaceutical Cocrystals with the Use of Laboratory X-ray Powder Diffraction Patterns. Crystals. 2023; 13(4):640. https://doi.org/10.3390/cryst13040640
Chicago/Turabian StyleChernyshev, Vladimir V. 2023. "Structural Characterization of Pharmaceutical Cocrystals with the Use of Laboratory X-ray Powder Diffraction Patterns" Crystals 13, no. 4: 640. https://doi.org/10.3390/cryst13040640
APA StyleChernyshev, V. V. (2023). Structural Characterization of Pharmaceutical Cocrystals with the Use of Laboratory X-ray Powder Diffraction Patterns. Crystals, 13(4), 640. https://doi.org/10.3390/cryst13040640