
Stereoselective Synthesis of New 4-Aryl-5-indolyl-1,2,4-triazole S- and N-β-Galactosides: Characterizations, X-ray Crystal Structure and Hirshfeld Surface Analysis

 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Instrumentation
2.2. General Procedures for Galactosylation
2.2.1. Method A
2.2.2. Method B
2.3. General Method for Thermal Transformation of S-Galactosides into N-Glycosides
Method C
2.4. Crystal Structure Determination
2.5. Hirshfeld Surface Analysis
3. Results and Discussion
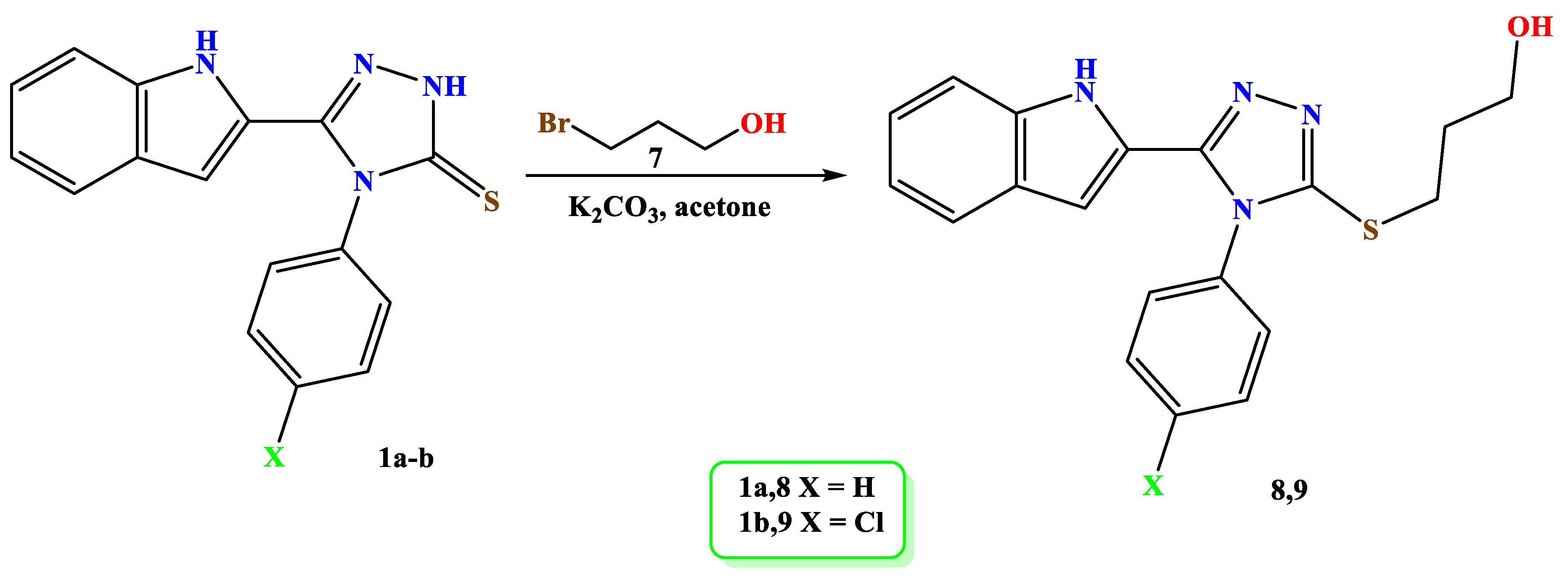
3.1. Chemistry
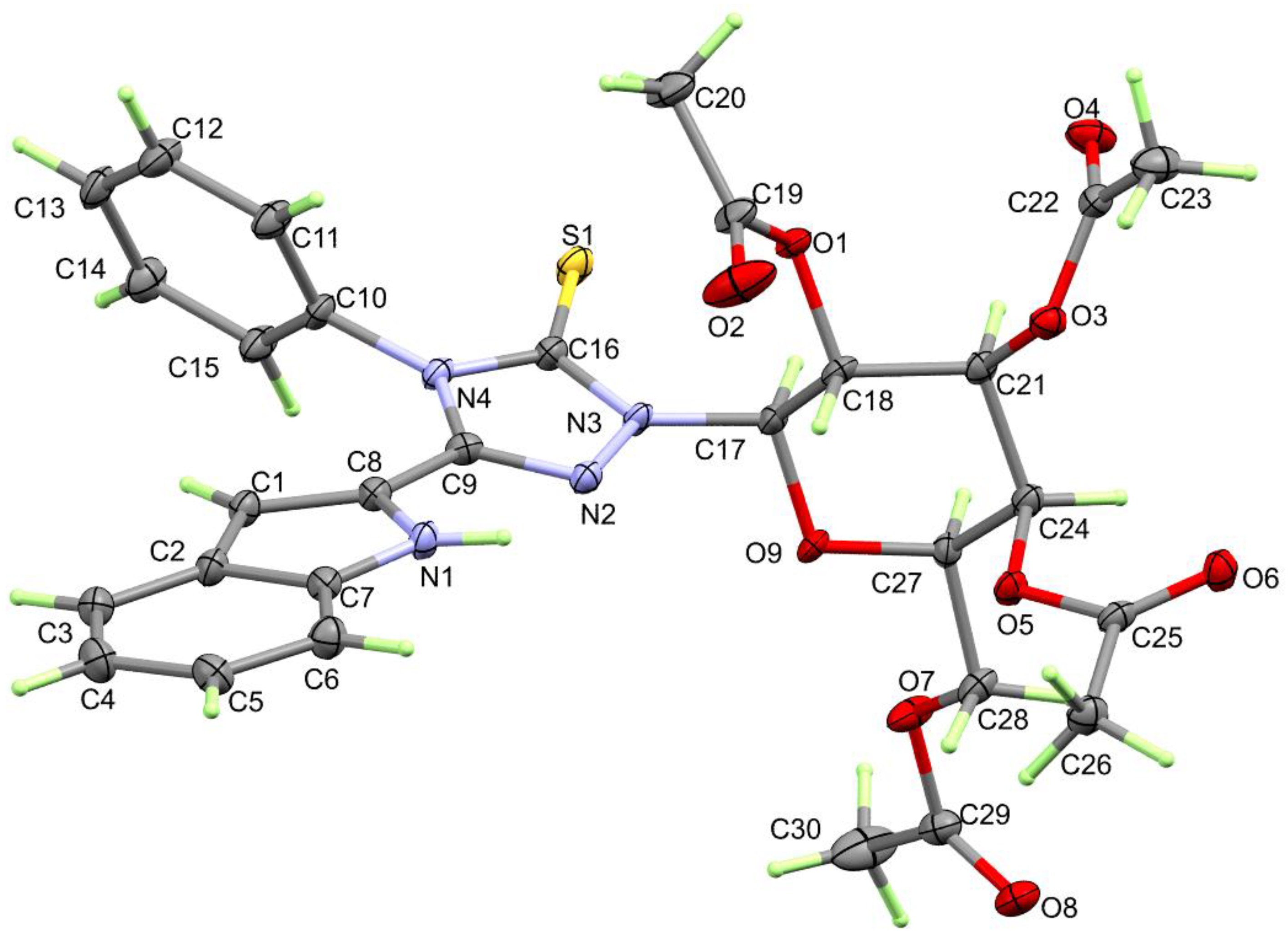
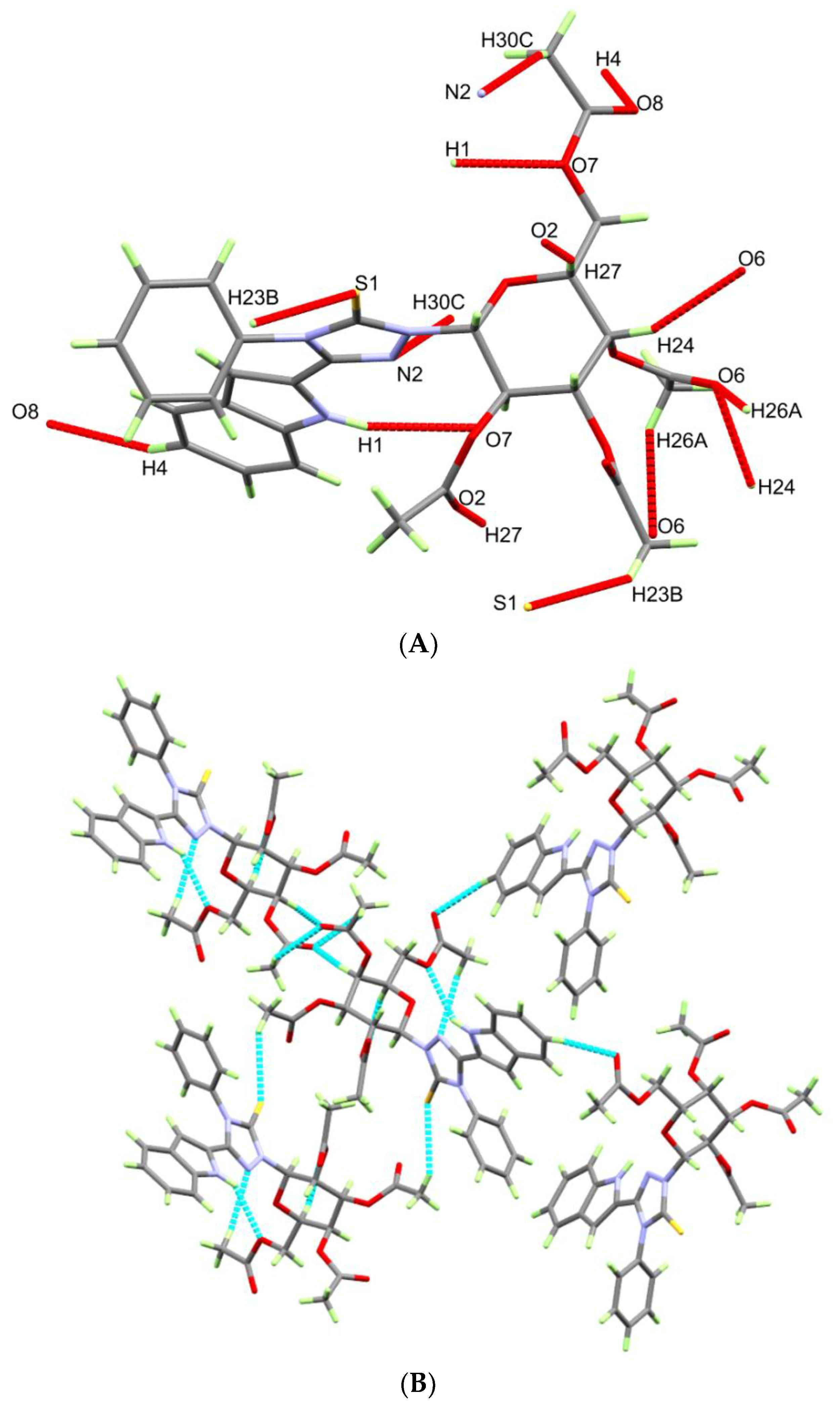
3.2. Crystal Structure Description
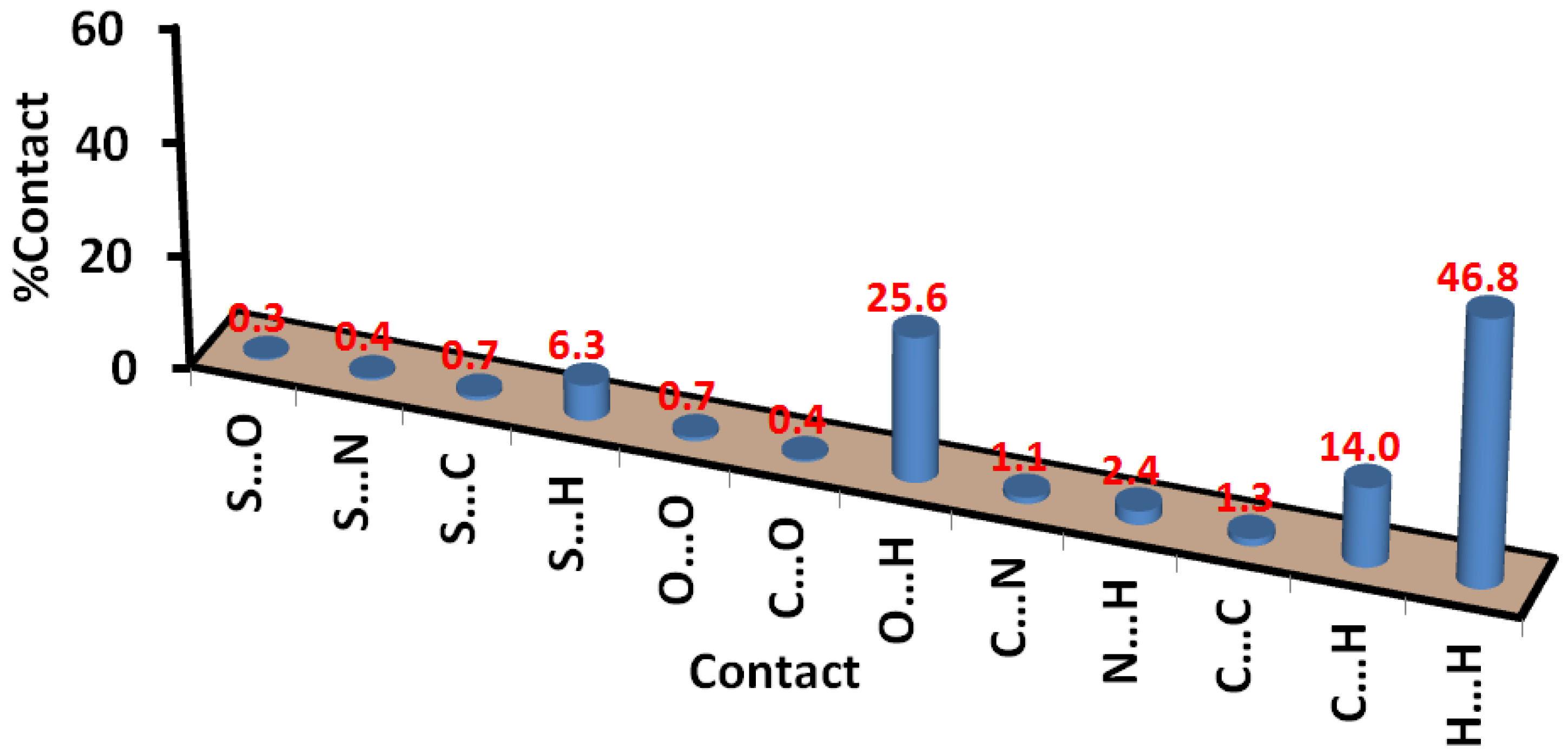
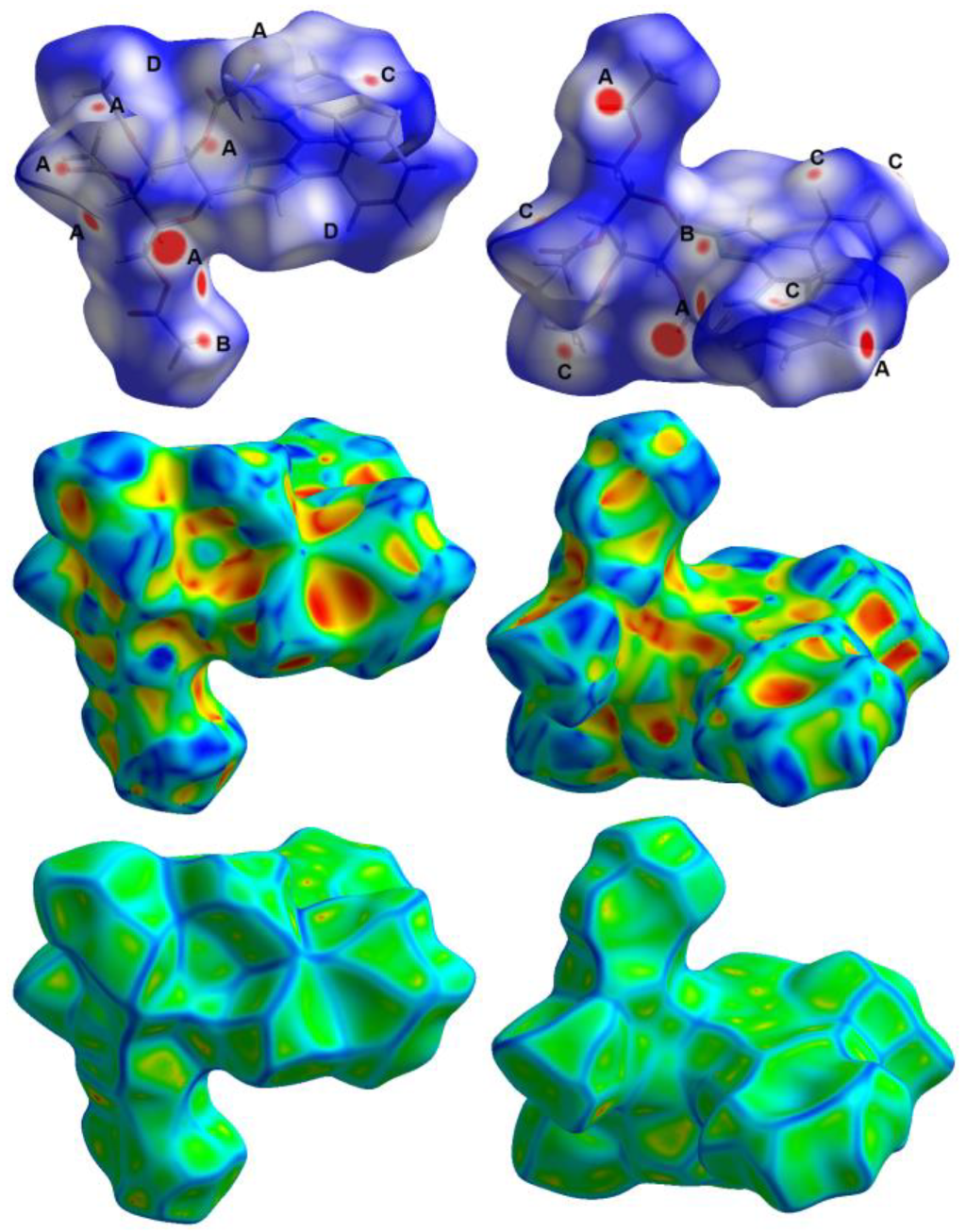
3.3. Hirshfeld Surface Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayes, M.R.; Pietruszka, J. Synthesis of Glycosides by Glycosynthases. Molecules 2020, 22, 1434. [Google Scholar] [CrossRef] [PubMed]
- Inman, M.; Moody, C.J. Indole synthesis-something old, something new. Chem. Sci. 2013, 4, 29–41. [Google Scholar] [CrossRef]
- Berger, M.; Gray, J.A.; Roth, B.L. The expanded biology of serotonin. Annu. Rev. Med. 2009, 60, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Kumar, P.; Pathaka, D. Biological importance of the indole nucleus in recent years: A comprehensive review. J. Heterocycl. Chem. 2010, 47, 491–502. [Google Scholar] [CrossRef]
- Zhang, M.Z.; Chen, Q.; Yang, G.F. A review on recent developments of indole-containing antiviral agents. Eur. J. Med. Chem. 2015, 89, 421–441. [Google Scholar] [CrossRef]
- Frost, J.M.; Dart, M.J.; Tietje, K.R.; Garrison, T.R.; Grayson, G.K.; Daza, A.V.; El-Kouhen, O.F.; Yao, B.B.; Hsieh, G.C.; Pai, M.; et al. Indol-3-ylcycloalkyl ketones: Effects of N1 substituted indole side chain variations on CB2 cannabinoid receptor activity. J. Med. Chem. 2010, 53, 295–315. [Google Scholar] [CrossRef]
- Rathod, A.S.; Godipurge, S.S.; Biradar, J.S. Microwave Assisted, Solvent-Free, “Green” Synthesis of Novel Indole Analogs as Potent Antitubercular and Antimicrobial Agents and Their Molecular Docking Studies. Russ. J. Gen. Chem. 2018, 88, 1238–1246. [Google Scholar] [CrossRef]
- Niemyjska, M.; MacIejewska, D.; Wolska, I.; Truszkowski, P. Synthesis, structural investigations, and anti-cancer activity of new methyl indole-3-carboxylate derivatives. J. Mol. Struct. 2012, 1026, 30–35. [Google Scholar] [CrossRef]
- Aggarwal, R.; Sumran, G. An Insight on Medicinal Attributes of 1,2,4-Triazoles. Eur. J. Med. Chem. 2020, 205, 112652. [Google Scholar] [CrossRef]
- Kaur, R.; Ranjan Dwivedi, A.; Kumar, B.; Kumar, V. Recent Developments on 1,2,4-Triazole Nucleus in Anticancer Compounds: A Review. Anti-Cancer Agents. Med. Chem. 2016, 16, 465–489. [Google Scholar]
- Wen, X.; Zhou, Y.; Zeng, J.; Liu, X. Recent Development of 1,2,4-Triazole-Containing Compounds as Anticancer Agents. Curr. Top. Med. Chem. 2020, 20, 1441–1460. [Google Scholar] [CrossRef] [PubMed]
- Shaker, R.M. The Chemistry of Mercapto- and Thione-Substituted 1,2,4-Triazoles and Their Utility in Heterocyclic Synthesis. Arkivoc 2006, 9, 59–112. [Google Scholar] [CrossRef]
- Slivka, M.V.; Korol, N.I.; Fizer, M.M. Fused Bicyclic 1,2,4-triazoles with One Extra Sulfur Atom: Synthesis, Properties, and Biological Activity. J. Heterocycl. Chem. 2020, 57, 3236–3254. [Google Scholar] [CrossRef]
- Küçükgüzel, Ş.G.; Çıkla-Süzgün, P. Recent Advances Bioactive 1,2,4-Triazole-3-Thiones. Eur. J. Med. Chem. 2015, 97, 830–870. [Google Scholar] [CrossRef]
- Patel, K.R.; Brahmbhatt, J.G.; Pandya, P.A.; Daraji, D.G.; Patel, H.D.; Rawal, R.M.; Baran, S.K. Design, Synthesis and Biological Evaluation of Novel 5-(4-Chlorophenyl)-4-Phenyl-4H-1,2,4-Triazole-3-Thiols as an Anticancer Agent. J. Mol. Struct. 2021, 1231, 130000. [Google Scholar] [CrossRef]
- Su, L.; Feng, Y.; Wei, K.; Xu, X.; Liu, R.; Chen, G. Carbohydrate-Based Macromolecular Biomaterials. Chem. Rev. 2021, 121, 10950–11029. [Google Scholar] [CrossRef]
- Borai, A.T.A.; Gomaa, M.S.; El Ashry, E.S.H.; Duerkop, A. Design, Synthesis, X-ray Crystal Structure and Regioselectivity Determination of Dihydro-Indolyl-1,2,4-triazole-3-thione and its 3-Benzylsulfanyl Analogues as Potent Anticancer Agents. Eur. J. Med. Chem. 2017, 125, 360–371. [Google Scholar] [CrossRef]
- Boraei, A.T.A.; Singh, P.K.; Sechi, M.; Satta, S. Discovery of novel functionalized 1,2,4-triazoles as PARP-1 inhibitors in breast cancer: Design, synthesis and antitumor activity evaluation. Eur. J. Med. Chem. 2019, 182, 111621. [Google Scholar] [CrossRef]
- Nafie, M.S.; Boraei, A.T.A. Exploration of novel VEGFR2 tyrosine kinase inhibitors via design and synthesis of new alkylated indolyl-triazole Schiff bases for targeting breast cancer. Bioorg. Chem. 2022, 122, 105708. [Google Scholar] [CrossRef]
- Youssef, M.F.; Nafie, M.S.; Salama, E.E.; Boraei, A.T.A.; Gad, E.M. Synthesis of New Bioactive Indolyl-1,2,4-Triazole Hybrids as Dual Inhibitors for EGFR/PARP-1 Targeting Breast and Liver Cancer Cells. ACS Omega 2022, 7, 45665–45677. [Google Scholar] [CrossRef]
- Al-Hussain, S.A.; Farghaly, T.A.; Zaki, M.E.A.; Abdulwahab, H.G.; Al-Qurashi, N.T.; Muhammad, Z.A. Discovery of novel indolyl-1,2,4-triazole hybrids as potent vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitors with potential anti-renal cancer activity. Bioorg. Chem. 2020, 105, 104330. [Google Scholar] [CrossRef] [PubMed]
- Maftei, C.V.; Fodor, E.; Jones, P.G.; Daniliuc, C.G.; Franz, M.H.; Kelter, G.; Fiebig, H.; Tamm, M.; Neda, I. Novel 1,2,4-oxadiazoles and trifluoromethylpyridines related to natural products: Synthesis, structural analysis and investigation of their antitumor activity. Tetrahedron 2016, 72, 1185–1199. [Google Scholar] [CrossRef]
- Neda, I.; Sakhaii, P.; Waßmann, A.; Niemeyer, U.; Günther, E.; Engel, J. A practical synthesis of benzyl α- and allyl β-D-glucopyranosides regioselectively substituted with (CH2)3OH groups: Stereocontrolled β-galactosidation by cation π-interaction. Synthesis 1999, 9, 1625–1632. [Google Scholar] [CrossRef]
- Rikagu Oxford Diffraction. CrysAlisPro; Rikagu Oxford Diffraction Inc.: Yarnton, UK, 2022. [Google Scholar]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H.J. OLEX2: A complete structure solution, refinement and analysis program. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer17 (2017) University of Western Australia. Available online: https://crystalexplorer.net/ (accessed on 30 July 2020).
- Panathur, N.; Dalimba, U.; Koushik, P.V.; Alvala, M.; Yogeeswari, P.; Sriram, D.; Kumar, V. Identification and characterization of novel indole based small molecules as anticancer agents through SIRT1 inhibition. Eur. J. Med. Chem. 2013, 69, 125–138. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 5 | 9 | |
|---|---|---|
| CCDC | 2243221 | 2243222 |
| empirical formula | C30H30N4O9S | C19H18.25ClN4O1.625S |
| fw | 622.64 | 396.13 |
| temp (K) | 120(2) K | 120(2) K |
| λ (Å) | 1.54184 Å | 1.54184 Å |
| cryst syst | orthorhombic | triclinic |
| space group | P212121 | P |
| a (Å) | a = 6.8865(3) Å | a = 11.0384(5) Å |
| b (Å) | b = 13.8302(6) Å | b = 13.6634(8) Å |
| c (Å) | c = 32.4134(19) Å | c = 14.0630(6) Å |
| α (deg) | 90 | 74.579(4)° |
| β (deg) | 90 | 74.477(4)° |
| γ(deg) | 90 | 72.100(5)° |
| V (Å3) | 3087.1(3) Å3 | 1905.31(18) Å3 |
| Z | 4 | 4 |
| ρcalc (Mg/m3) | 1.340 Mg/m3 | 1.381 Mg/m3 |
| μ(Mo Kα) (mm−1) | 1.440 mm−1 | 2.964 mm−1 |
| no. reflns. | 12175 | 33118 |
| unique reflns. | 6313 | 7107 |
| completeness to θ=67.684° | 100% | 99.9% |
| GOOF (F2) | 1.029 | 1.057 |
| Rint | 0.0633 | 0.0402 |
| R1a (I ≥ 2σ) | 0.0563 | 0.0489 |
| wR2b (I ≥ 2σ) | 0.1123 | 0.1378 |
| Bond | Length/Å | Bond | Length/Å |
|---|---|---|---|
| 5 | 9 | ||
| S(1)-C(16) | 1.654(5) | Cl(1)–C(13) | 1.741(3) |
| O(1)-C(19) | 1.357(6) | N(15)–C(1) | 1.371(3) |
| O(1)-C(18) | 1.442(6) | N(15)–C(8) | 1.382(3) |
| N(1)-C(7) | 1.380(7) | N(16)–C(16) | 1.370(3) |
| N(1)-C(8) | 1.385(7) | N(16)–C(9) | 1.377(3) |
| O(2)-C(19) | 1.188(7) | N(16)–C(10) | 1.444(3) |
| N(2)-C(9) | 1.311(7) | N(17)–C(16) | 1.315(3) |
| N(2)-N(3) | 1.393(6) | N(17)–N(18) | 1.399(3) |
| O(3)-C(22) | 1.367(7) | N(18)–C(9) | 1.311(3) |
| O(3)-C(21) | 1.429(6) | C(16)–S(1) | 1.732(3) |
| D-H...A | d(D-H) | d(H...A) | d(D...A) | <(DHA) | Symm. Code |
|---|---|---|---|---|---|
| N(1)-H(1)...O(7) | 0.85(6) | 2.48(6) | 3.238(6) | 149(5) | x − 1, y, z |
| C(23)-H(23B)...S(1) | 0.98 | 2.98 | 3.821(6) | 144.8 | x − 1/2, −y + 1/2, −z + 1 |
| C(24)-H(24)...O(6) | 1.00 | 2.47 | 3.416(6) | 157.1 | x + 1/2, −y + 3/2, −z + 1 |
| C(26)-H(26A)...O(6) | 0.98 | 2.56 | 3.290(7) | 131 | x − 1/2, −y + 3/2, −z + 1 |
| C(27)-H(27)...O(2) | 1.00 | 2.21 | 3.177(7) | 163.4 | x + 1, y, z |
| C(30)-H(30C)...N(2) | 0.98 | 2.61 | 3.531(8) | 156.2 | x + 1, y, z |
| Contact | Distance | Contact | Distance |
|---|---|---|---|
| H1…O7 | 2.607 | H14…C7 | 2.784 |
| H6…O7 | 2.348 | H14…C6 | 2.737 |
| H24…O6 | 2.396 | H14…C5 | 2.733 |
| H26A…O6 | 2.497 | H15…C3 | 2.685 |
| H26C…O8 | 2.606 | H24…C25 | 2.767 |
| H27…O2 | 2.126 | H23A…C25 | 2.666 |
| H30C…N2 | 2.518 | H23A…C26 | 2.771 |
| H11…S1 | 3.025 | H24…C25 | 2.767 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altowyan, M.S.; Haukka, M.; Soliman, S.M.; Barakat, A.; Boraei, A.T.A.; Aboelmagd, A. Stereoselective Synthesis of New 4-Aryl-5-indolyl-1,2,4-triazole S- and N-β-Galactosides: Characterizations, X-ray Crystal Structure and Hirshfeld Surface Analysis. Crystals 2023, 13, 797. https://doi.org/10.3390/cryst13050797
Altowyan MS, Haukka M, Soliman SM, Barakat A, Boraei ATA, Aboelmagd A. Stereoselective Synthesis of New 4-Aryl-5-indolyl-1,2,4-triazole S- and N-β-Galactosides: Characterizations, X-ray Crystal Structure and Hirshfeld Surface Analysis. Crystals. 2023; 13(5):797. https://doi.org/10.3390/cryst13050797
Chicago/Turabian StyleAltowyan, Mezna Saleh, Matti Haukka, Saied M. Soliman, Assem Barakat, Ahmed T. A. Boraei, and Ahmed Aboelmagd. 2023. "Stereoselective Synthesis of New 4-Aryl-5-indolyl-1,2,4-triazole S- and N-β-Galactosides: Characterizations, X-ray Crystal Structure and Hirshfeld Surface Analysis" Crystals 13, no. 5: 797. https://doi.org/10.3390/cryst13050797