
Structure, Conformation and Contact Analyses of Six Aromatic Diamide Diesters


Abstract
:1. Introduction
2. Experimental
2.1. Materials and Characterisation
2.2. Synthetic Reaction Procedure and Characterisation
Reaction Procedures
2.3. Spectral Data and Characterisation
3. Results and Discussion
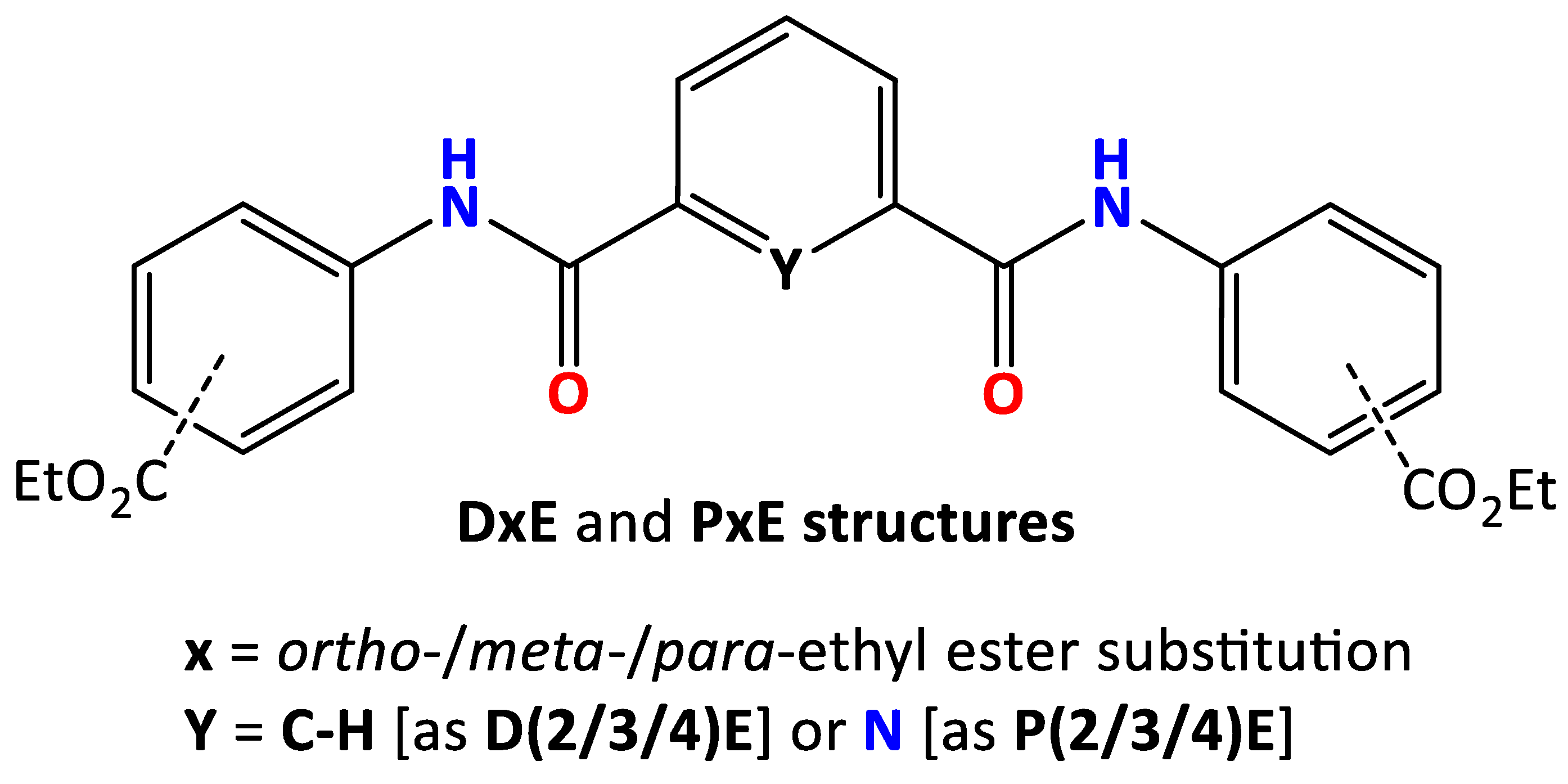
3.1. Crystal and Molecular Structural Analyses of the six PxE and DxE
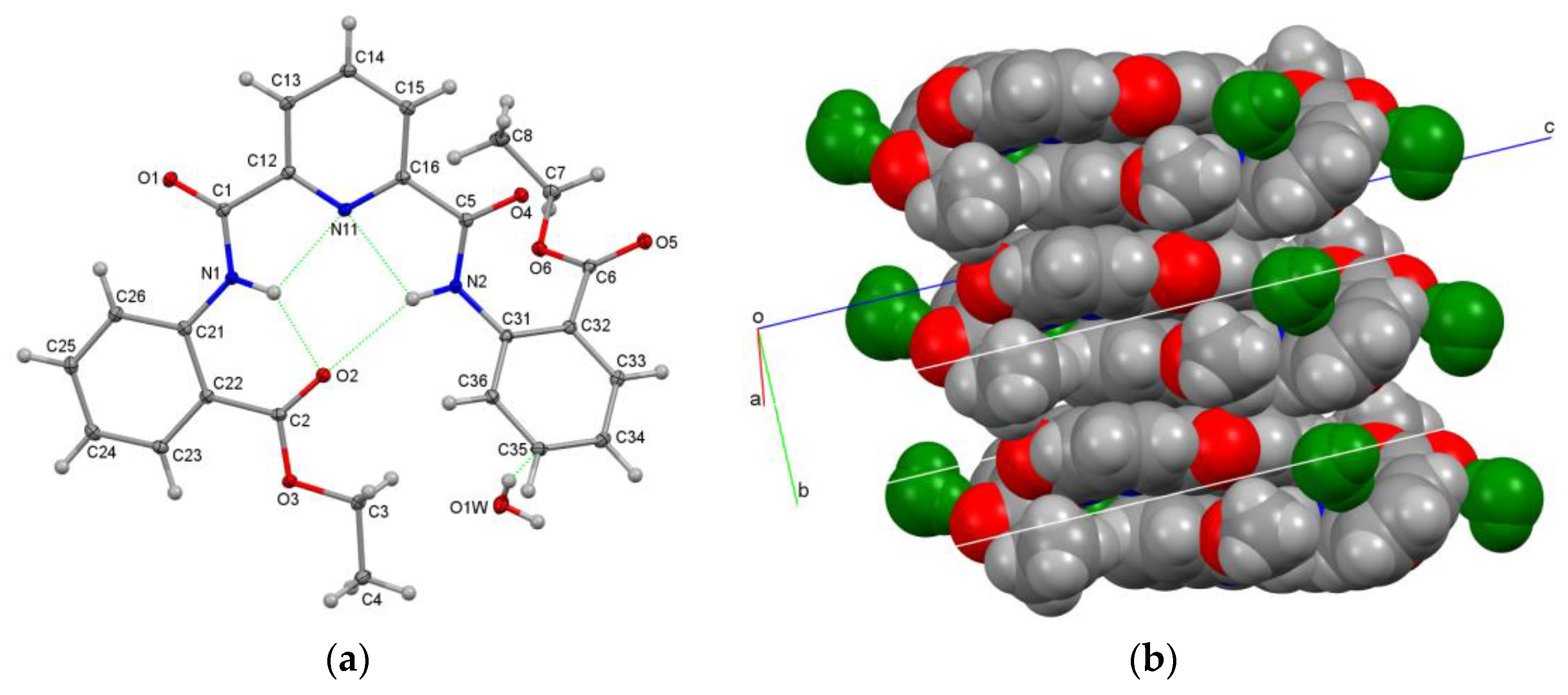
3.2. The Three DxE Isophthalamide Diesters’ Crystal Structures
3.2.1. Consequences of the Hierarchical Nature of Interactions and Molecular Twinning
3.2.2. The Planar 34-Nonhydrogen-Atom Molecular Structure of D2E
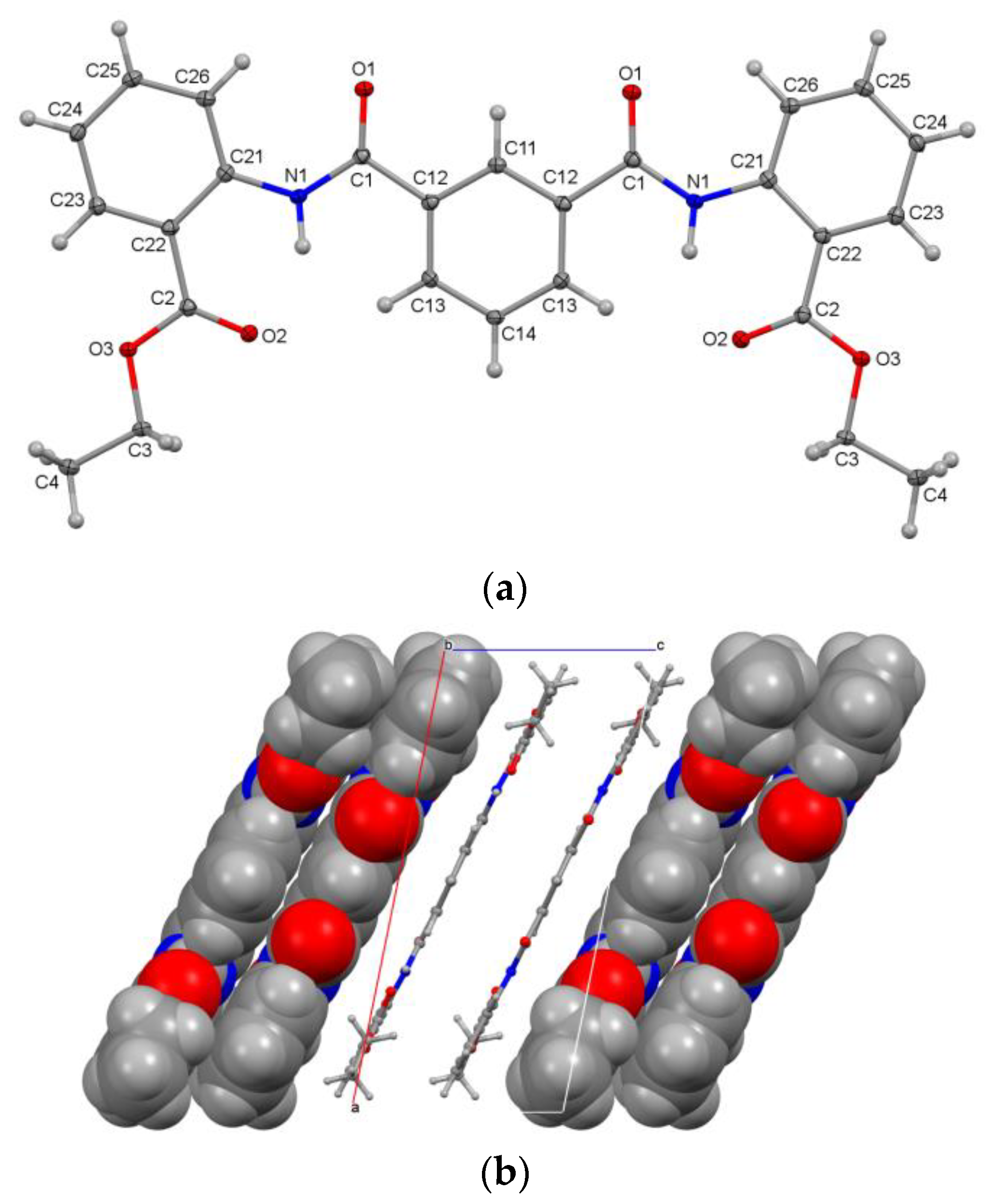
3.2.3. Aggregation of Molecules A–D in D3E
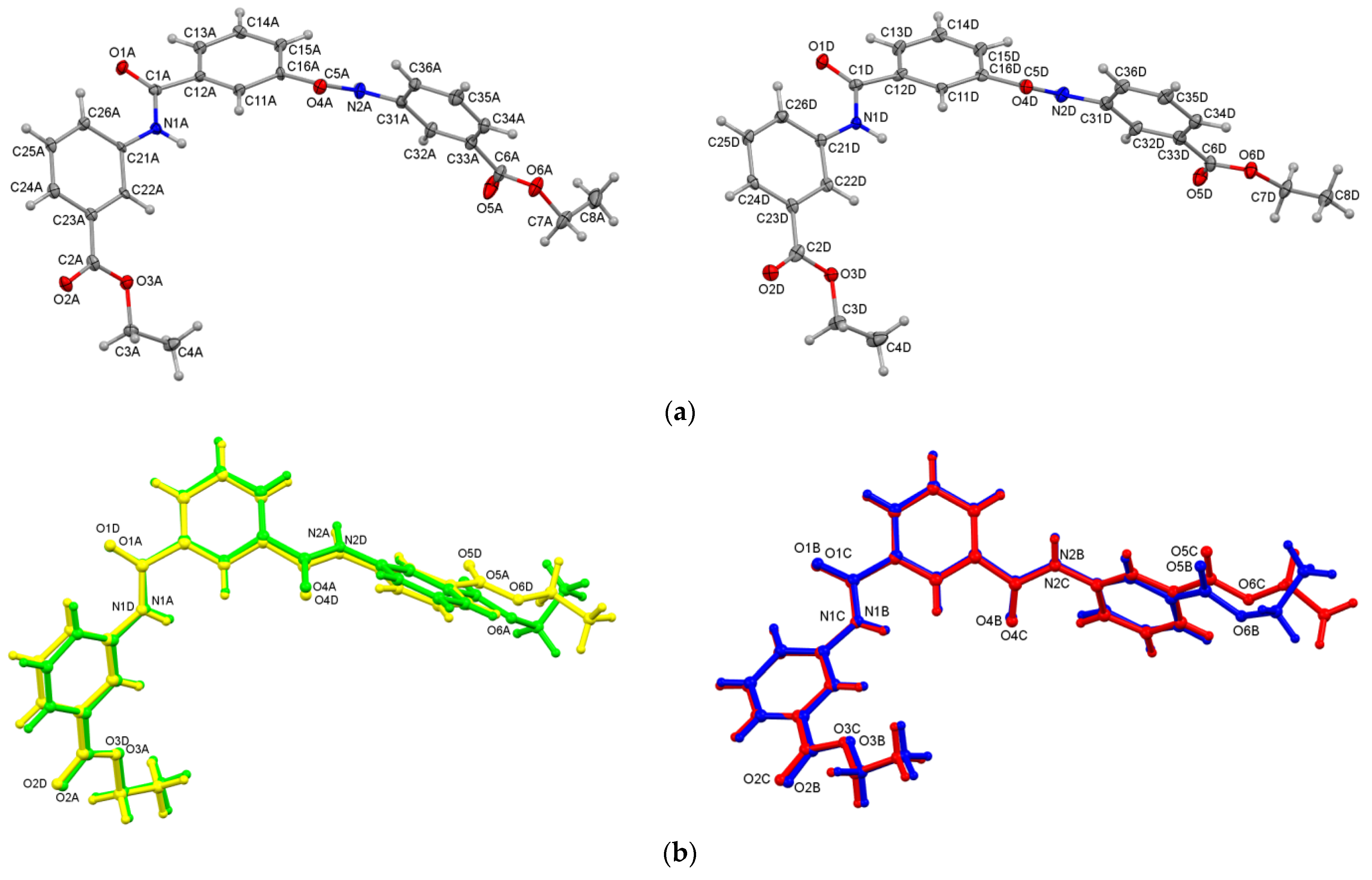
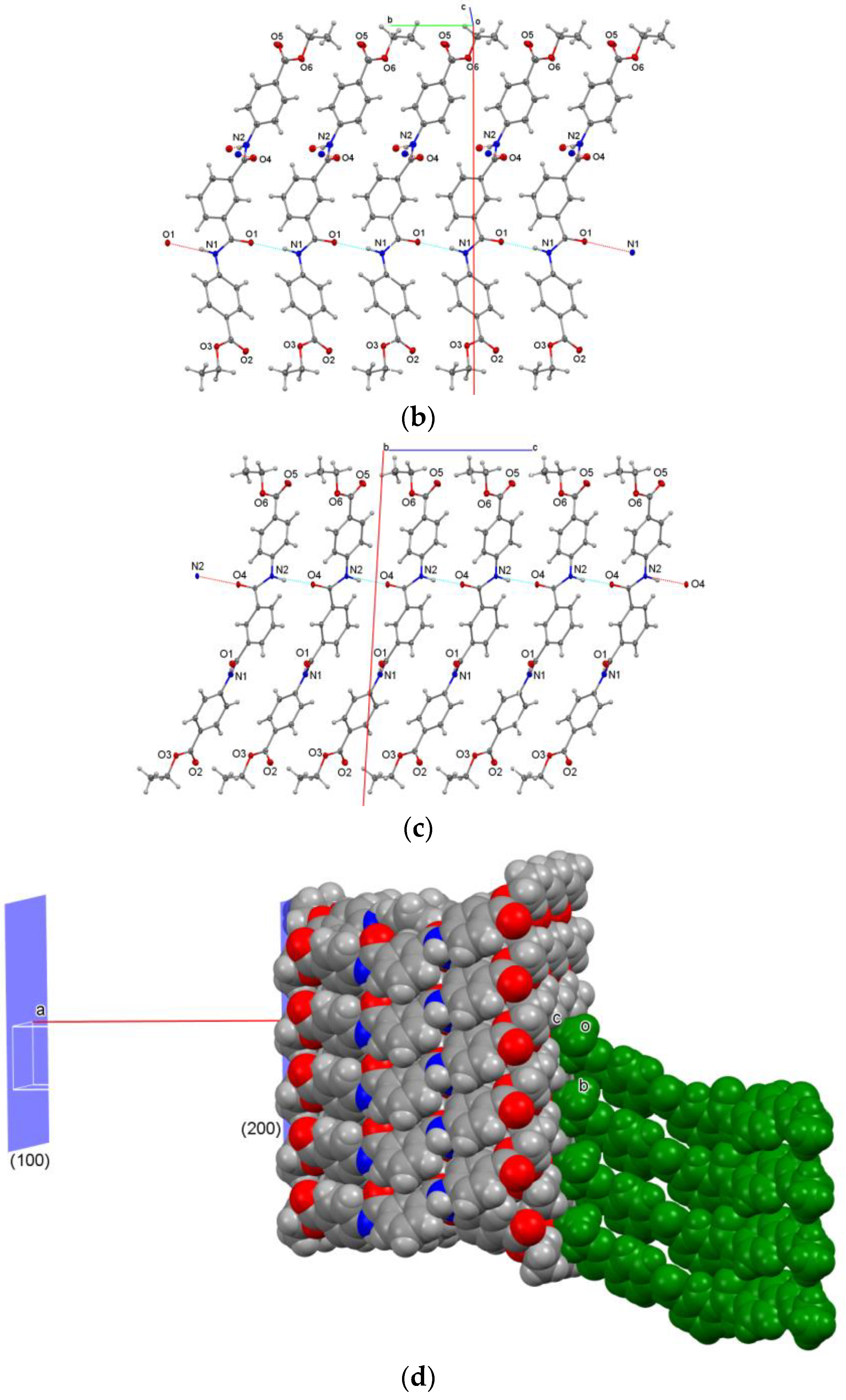
3.2.4. Twinning and Sheet Formation in D4E
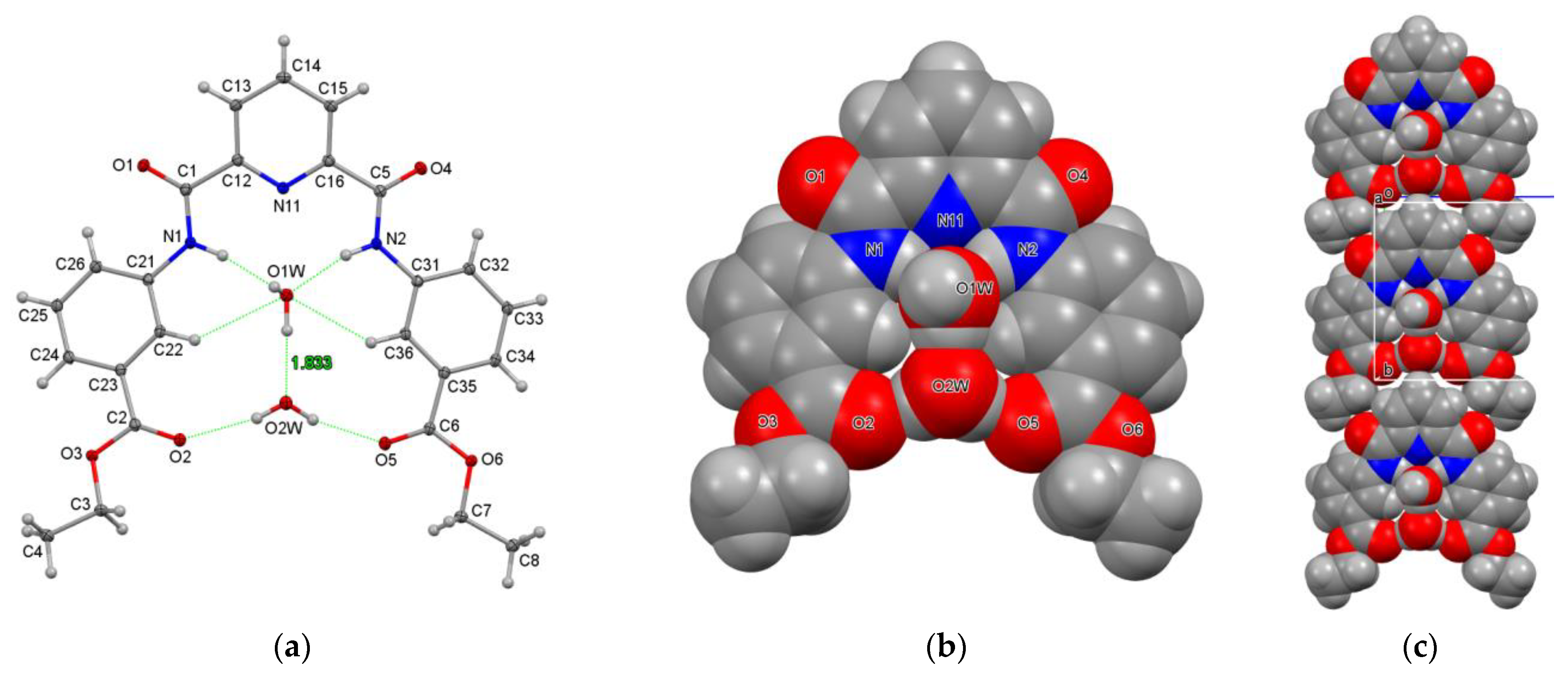
3.3. The Three PxE Pyridinedicarboxamide Ester Structures
3.3.1. Compact Aromatic Ring⋯Ring Stacking in the Crystal Structure of P2E·0.441(H2O)
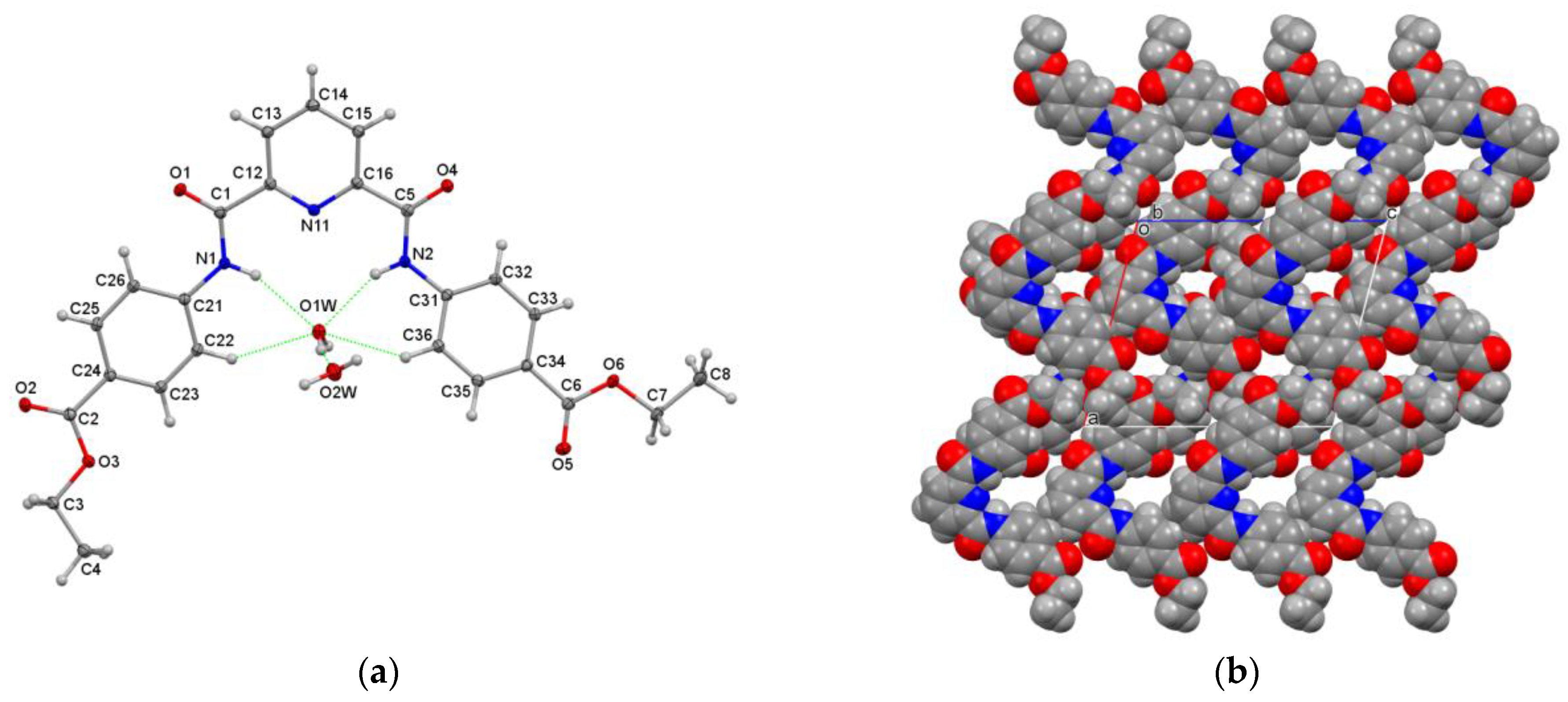
3.3.2. Hydrogen-Bonded Water Molecules in Niches of P3E·2H2O
3.3.3. Hydrogen-Bonded Water Molecules in P4E·2H2O
3.4. Hirshfeld Surface Analysis
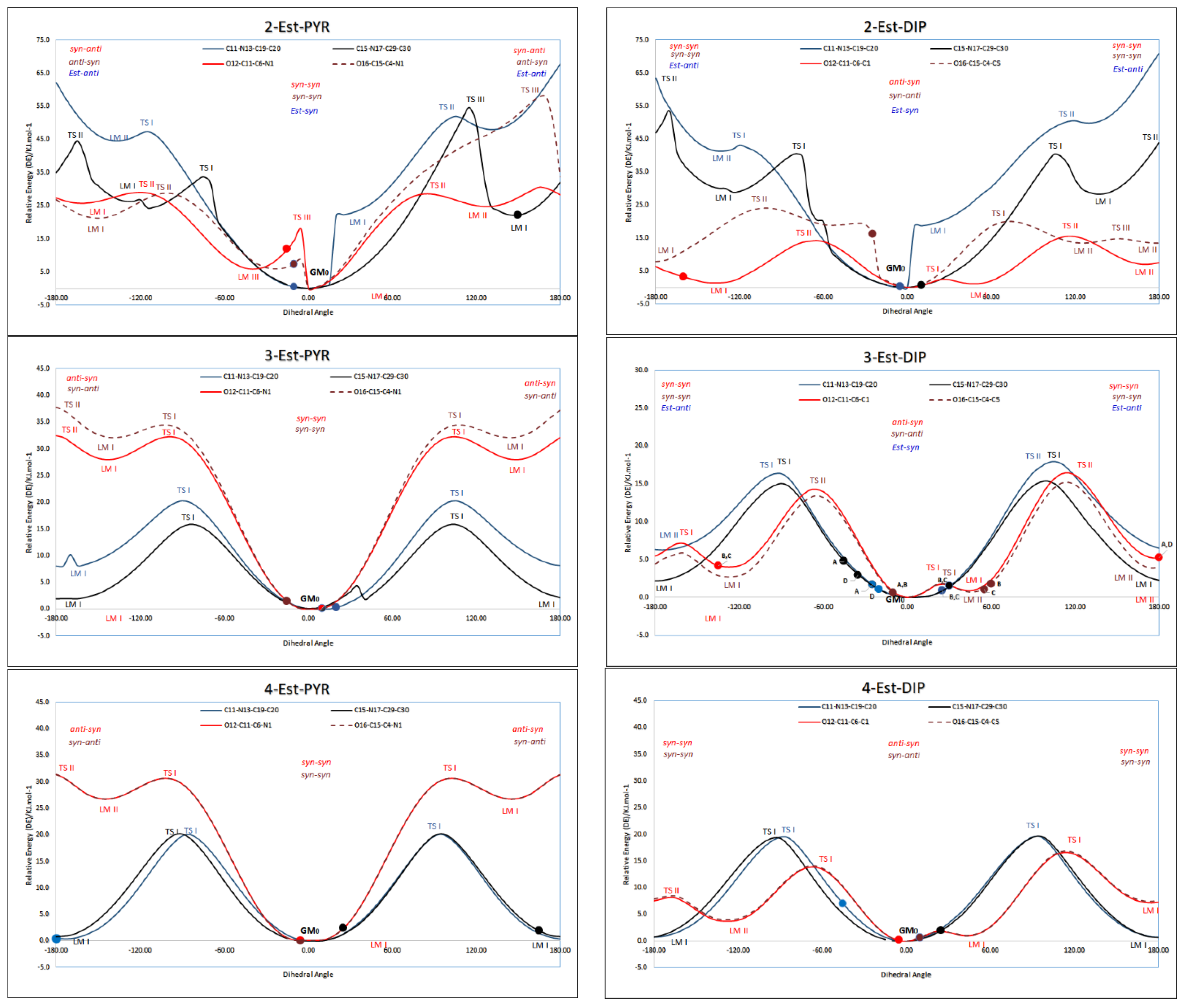
3.5. Conformational Analysis
3.5.1. Structure Optimisation
3.5.2. Conformational Analysis
3.6. Related Research Work
4. Conclusions and Future Work
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Greenberg, A.; Breneman, C.M.; Liebman, J.F. (Eds.) The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science; Wiley: New York, NY, USA, 2000. [Google Scholar]
- Lappert, M.F.; Protchenko, A.V.; Power, P.P.; Seeber, A.L. Metal Amide Chemistry; John Wiley: Hoboken, NJ, USA, 2008. [Google Scholar]
- Szostak, M. Amide Bond Activation. Molecules 2019. [Google Scholar] [CrossRef]
- Meng, G.; Zhang, J.; Szostak, M. Acyclic Twisted Amides. Chem. Rev. 2021, 121, 12746–12783. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Higson, S.; Davis, F. Macrocycles: Construction, Chemistry and Nanotechnology Applications; John Wiley & Sons Ltd.: Chichester, UK, 2011. [Google Scholar]
- Tranchemontagne, D.J.; Mendoza-Cortés, J.L.; O’Keeffe, M.; Yaghi, O.M. Secondary building units, nets and bonding in the chemistry of metal–organic frameworks. Chem. Soc. Rev. 2009, 38, 1257–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Chen, Y.-P.; Perry, Z.; Li, J.-R.; Zhou, H.-C. Preparation of Core–Shell Coordination Molecular Assemblies via the Enrichment of Structure-Directing “Codes” of Bridging Ligands and Metathesis of Metal Units. J. Am. Chem. Soc. 2014, 136, 16895–16901. [Google Scholar] [CrossRef]
- Abdolmaleki, S.; Ghadermazi, M. Novel pyridinedicarboxamide derivatives and a polymeric copper(II) complex: Synthesis, structural characterization, electrochemical behavior, catalytic and cytotoxic studies. Inorg. Chim. Acta 2017, 461, 221–232. [Google Scholar] [CrossRef]
- Karmakar, A.; Rúbio, G.M.D.M.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Synthesis of Metallomacrocycle and Coordination Polymers with Pyridine-Based Amidocarboxylate Ligands and Their Catalytic Activities towards the Henry and Knoevenagel Reactions. ChemistryOpen 2018, 7, 865–877. [Google Scholar] [CrossRef]
- Howlader, P.; Mukherjee, P.S. Solvent Directed Synthesis of Molecular Cage and Metal Organic Framework of Copper(II) Paddlewheel Cluster. Isr. J. Chem. 2019, 59, 292–298. [Google Scholar] [CrossRef]
- Karmakar, A.; Soliman, M.M.A.; Rúbio, G.M.D.M.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Synthesis and catalytic activities of a Zn(ii) based metallomacrocycle and a metal–organic framework towards one-pot deacetalization-Knoevenagel tandem reactions under different strategies: A comparative study. Dalton Trans. 2020, 49, 8075–8085. [Google Scholar] [CrossRef]
- Gallagher, J.F.; Sheehy, M.J.; Kenny, P.T.M. Synthesis and electrochemical anion recognition by novel redox-responsive ferrocenoyl dipeptide ester derivatives; 1H NMR anion complexation studies. Inorg. Chem. Commun. 1999, 8, 327–330. [Google Scholar] [CrossRef]
- Savage, D.; Kenny, P.T.M.; Gallagher, J.F.; Ida, Y. Synthesis and structural characterization of N-para-ferrocenyl benzoyl amino acid ethyl esters and the X-ray crystal structures of the glycyl and (±)-2-aminobutyric acid derivative Fc-C6H4CONHCH(C2H5)CO2Et. Inorg. Chem. Commun. 2004, 5, 1034–1040. [Google Scholar] [CrossRef]
- Gallagher, J.F.; Alley, S.; Brosnan, M.; Lough, A.J. 1,1′-Fc(4-C6H4CO2Et)2 and its unusual salt derivative with Z′ = 5, catena-[Na+]2[1,1′-Fc(4-C6H4CO2−)2]·0.6H2O [1,1′-Fc = (η5-(C5H4)2Fe]. Acta Crystallogr. 2010, B66, 196–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, J.F.; Alley, S.; Lough, A.J. A structural systematic study of semi-rigid ferrocene derivatives as a 3 × 3 metallocene isomer grid: P-/m-/o-(FcC6H4)CONH(p-/m-/o-C6H4)CO2Et, [Fc = (η5-C5H5)Fe(η5-C5H4)]. Inorg. Chim Acta 2016, 444, 113–125. [Google Scholar] [CrossRef]
- Mocilac, P.; Gallagher, J.F. Entry point into new trimeric and tetrameric imide-based macrocyclic esters derived from isophthaloyl dichloride and methyl 6-aminonicotinate. Acta Crystallogr. 2012, B69, 62–69. [Google Scholar] [CrossRef]
- Osman, I.A.; Mocilac, P.; Gallagher, J.F. Short C–H⋯F interactions involving the 2,5-difluorobenzene group: Understanding the role of fluorine in aggregation and complex C–F/C–H disorder in a 2 × 6 isomer grid. CrystEngComm 2016, 18, 5764–5776. [Google Scholar]
- Osman, I.A.; McKee, V.; Jelsch, C.; Gallagher, J.F. Roles of Hydrogen, Halogen bonding and Aromatic stacking in a series of isophthalamides. Symmetry 2023, 15, 738. [Google Scholar] [CrossRef]
- Møller, M.S.; Liljedahl, M.C.; McKee, V.; McKenzie, C.J. Solid phase nitrosylation of enantiomeric cobalt(II) complexes. Chemistry 2021, 3, 585–597. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09 Revis. B.01; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Becke, A.D. Density-Functional Thermochemistry. 3. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. 20. Basis Set for Correlated Wave-Functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Guillot, B.; Enrique, E.; Huder, L.; Jelsch, C. MoProViewer: A tool to study proteins from a charge density science perspective. Acta Crystallogr. 2014, A70, C279. [Google Scholar] [CrossRef]
- Jelsch, C.; Ejsmont, K.; Huder, L. The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis. IUCrJ 2014, 1, 119–128. [Google Scholar] [CrossRef]
- Nowak-Król, A.; Würthner, F. Progress in the synthesis of perylene bisimide dyes. Org. Chem. Front. 2019, 6, 1272–1318. [Google Scholar] [CrossRef] [Green Version]
- Maeda, T.; Liess, A.; Kudzus, A.; Krause, A.-M.; Stolte, M.; Amitani, H.; Yagi, S.; Fujiwara, H.; Würthner, F. Hydrogen bond-rigidified planar squaraine dye and its electronic and organic semiconductor properties. Chem. Commun. 2020, 56, 9890–9893. [Google Scholar] [CrossRef]
- Gorelik, T.E.; Bekő, S.L.; Teteruk, J.; Heyse, W.; Schmidt, M.U. Analysis of diffuse scattering in electron diffraction data for the crystal structure determination of Pigment Orange 13, C32H24Cl2N8O2. Acta Crystallogr. 2023, B77, 122–137. [Google Scholar] [CrossRef]
- Baggio, R. Playing around with MP, a tool for the analysis of pseudosymmetry: Recurrent appearance of local pseudo-space groups in the asymmetric unit of Z’ = 4 structures. Acta Crystallogr. 2020, B76, 258–268. [Google Scholar] [CrossRef]
- Brock, C.P.; Taylor, R. Identifying and characterizing translationally modulated molecular crystal structures. Acta Crystallogr. 2020, B76, 630–634. [Google Scholar] [CrossRef]
- Reddy, C.M.; Gundakaram, R.C.; Basavoju, S.; Kirchner, M.T.; Padmanabhan, K.A.; Desiraju, G.R. Structural basis for bending of organic crystals. Chem. Commun. 2005, 31, 3945–3947. [Google Scholar] [CrossRef]
- Takamizawa, S.; Takasaki, Y. Superelastic Shape Recovery of Mechanically Twinned 3,5-Difluorobenzoic Acid Crystals. Angew. Chem. Int. Ed. 2015, 54, 4815–4817. [Google Scholar] [CrossRef]
- Ferguson, G.; Gallagher, J.F.; Glidewell, C.; Zakaria, C.M. O—H⋯π(arene) Intermolecular Hydrogen Bonding in the Structure of 1,1,2-Triphenylethanol. Acta Crystallogr. 1994, C50, 70–73. [Google Scholar] [CrossRef] [Green Version]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Malone, J.F.; Murray, C.M.; Dolan, G.M.; Docherty, R.; Lavery, A.J. Intermolecular Interactions in the Crystal Chemistry of N,N′-Diphenylisophthalamide, Pyridine-2,6-dicarboxylic Acid Bisphenylamide, and Related Compounds. Chem. Mater. 1997, 9, 2983–2989. [Google Scholar] [CrossRef]
- Picci, G.; Bazzicalupi, C.; Coles, S.J.; Gratteri, P.; Isaia, F.; Lippolis, V.; Montis, R.; Murgia, S.; Nocentini, A.; Orton, J.B.; et al. Halogenated isophthalamides and dipicolineamides: The role of the halogen substituents in the anion binding properties. Dalton Trans. 2020, 49, 9231–9238. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, J.F.; Ferguson, G.; McAlees, A. Ligands for application in Coordination Chemistry: Three Dicarboxylic Acids. Acta Crystallogr. 1995, C51, 454–458. [Google Scholar]
- Atcher, J.; Mateus, P.; Kauffmann, B.; Rosu, F.; Maurizot, V.; Huc, I. Large-Amplitude Conformational Changes in Self-Assembled Multi-Stranded Aromatic Sheets. Angew. Chem. Int. Ed. 2021, 60, 2574–2577. [Google Scholar] [CrossRef]
- Bindl, D.; Mandal, P.K.; Huc, I. Generalizing the aromatic δ-Amino Acid Foldamer Helix. Chem. A Eur. J. 2022, 28, e202200538. [Google Scholar] [CrossRef]
- Montis, R.; Fusaro, L.; Falqui, A.; Hursthouse, M.B.; Tumanov, N.; Coles, S.J.; Threlfall, T.L.; Horton, P.N.; Sougrat, R.; Lafontaine, A.; et al. Complex structures arising from self-assembly of a simple organic salt. Nature 2021, 590, 275–278. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structures (DxE Are Twinned) | Crystal System, Space Group | Z′ | Volume (Å3) | R, wR2 R-Factors, GoF |
|---|---|---|---|---|
| D2E | Monoclinic, C2/c | 0.5 | 2155.4(4) | 0.032, 0.070, 0.814 |
| D3E | Monoclinic, P21 | 4 | 4467.8(2) | 0.077, 0.207, 1.022 |
| D4E | Monoclinic, P21/c | 1 | 2230.76(8) | 0.079, 0.186, 1.101 |
| P2E·0.441(H2O) | Monoclinic, P21/n | 1 | 2176.60(6) | 0.031, 0.080, 1.054 |
| P3E·2(H2O) | Monoclinic, I2/a | 1 | 4673.65(9) | 0.030, 0.082, 1.025 |
| P4E·2(H2O) | Monoclinic, P21/c | 1 | 2325.43(6) | 0.039, 0.110, 1.022 |
| Structure | C6/C6 DxE or C5N/C6 PxE | centC6/amide | termC6/amide | N⋯N/O | Primary Packing |
|---|---|---|---|---|---|
| D2E | 0.14(7) 3.33(8) | 2.88(7) | 1.39(6) | 2.6509(17) | Ring⋯ring stacking |
| D3E Mols (A–D) | 40.4(2) to 47.5(2) 62.52(19) to 71.08(19) 87.13(17) to 89.93(18) | 23.1(3), 37.1(2) 22.9(3), 37.2(2) 22.5(3), 34.3(3) 23.5(3), 34.5(3) | 24.5(4), 34.2(4) 20.5(4), 32.5(3) 21.5(4), 28.7(3) 17.0(4), 28.1(3) | 3.034(9), 3.065(9) 3.022(9), 3.032(9) 2.962(9), 3.004(10) 2.982(9), 3.007(9) | Amide⋯amide |
| D4E | 3.4(2) 73.41(9) 75.71(10) | 27.76(18) 43.25(13) | 30.23(15) 31.11(16) | 2.992(4) 2.899(4) | Amide⋯amide |
| P2E ·0.441(H2O) | 2.55(5) 41.65(3) | 3.88(5) 43.66(3) | 5.23(5) 2.07(5) | 2.6548(12)# 2.6443(13) 3.1609(12)# 2.829(2) | Intramolecular O1W⋯O=C |
| P3E·2(H2O) | 20.00(2) 14.43(2) | 14.67(3) 3.44(2) | 5.38(2) 11.59(2) | 3.0348(11) 3.0001(11) | 2D sheets |
| P4E·2(H2O) | 11.58(3) 24.65(3) | 8.51(2) 20.17(3) | 3.24(3) 4.60(3) | 3.0900(17) 2.9639(17) | 2D sheets |
| Dihedral Angle | α1 | β1 | γ1 | α2 | β2 | γ2 | Energy |
|---|---|---|---|---|---|---|---|
| D2E | −160.79 | 4.71 | −175.45 | −153.93 | −128.92 | −159.49 | −4115.33 |
| D3E | −148.90 | 5.30 | −170.94 | −149.06 | 6.48 | −170.96 | −4115.73 |
| D4E | −148.05 | 6.34 | −170.47 | −148.00 | 6.70 | −170.54 | −4115.75 |
| P2E | −165.68 | 20.24 | −175.12 | −173.70 | −128.09 | −172.57 | −4157.47 |
| P3E | 179.99 | 0.0003 | 180.0 | 179.99 | −0.004 | 179.99 | −4157.933 |
| P4E | −177.82 | 0.78 | −179.70 | −177.44 | 0.78 | −179.64 | −4157.934 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osman, I.A.; McKee, V.; Jelsch, C.; Gallagher, J.F. Structure, Conformation and Contact Analyses of Six Aromatic Diamide Diesters. Crystals 2023, 13, 1133. https://doi.org/10.3390/cryst13071133
Osman IA, McKee V, Jelsch C, Gallagher JF. Structure, Conformation and Contact Analyses of Six Aromatic Diamide Diesters. Crystals. 2023; 13(7):1133. https://doi.org/10.3390/cryst13071133
Chicago/Turabian StyleOsman, Islam Ali, Vickie McKee, Christian Jelsch, and John F. Gallagher. 2023. "Structure, Conformation and Contact Analyses of Six Aromatic Diamide Diesters" Crystals 13, no. 7: 1133. https://doi.org/10.3390/cryst13071133
APA StyleOsman, I. A., McKee, V., Jelsch, C., & Gallagher, J. F. (2023). Structure, Conformation and Contact Analyses of Six Aromatic Diamide Diesters. Crystals, 13(7), 1133. https://doi.org/10.3390/cryst13071133