
Isocyanide π-Hole Interactions Supported by Aurophilic Forces

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents, Instrumentation, and Methods
2.2. X-ray Diffraction Study
2.3. Computational Details
3. Results and Discussion
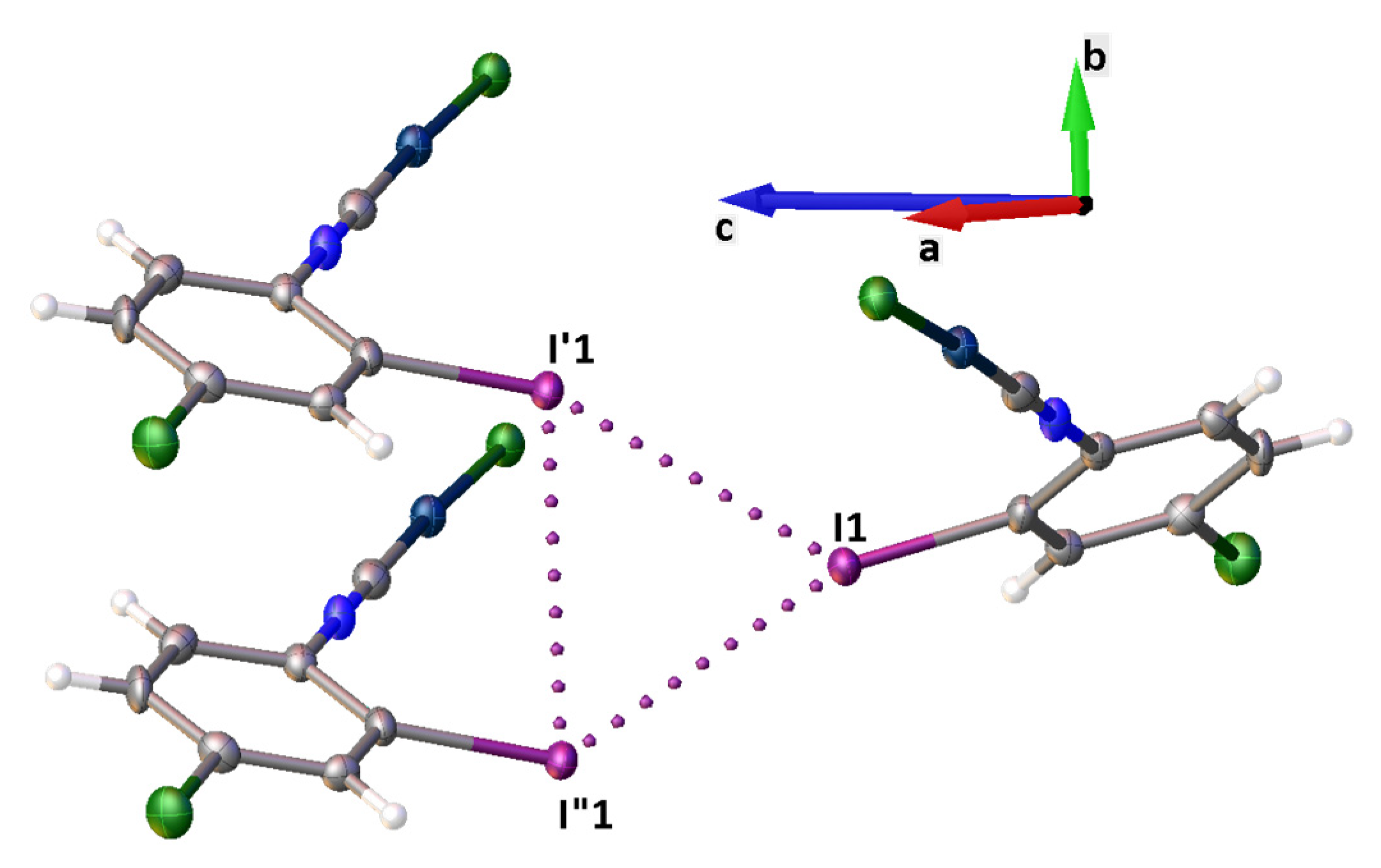
3.1. X-ray Diffraction Study
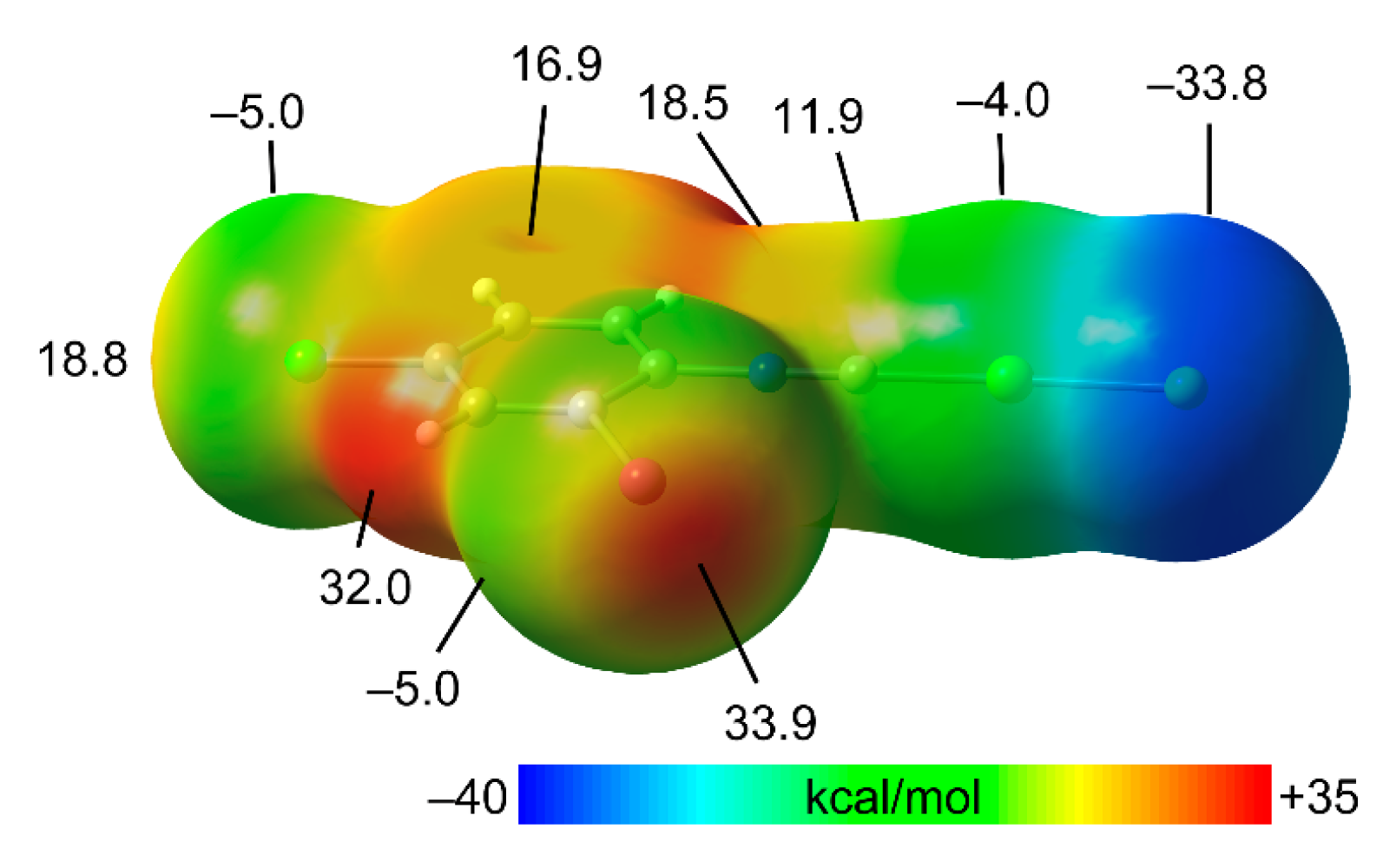
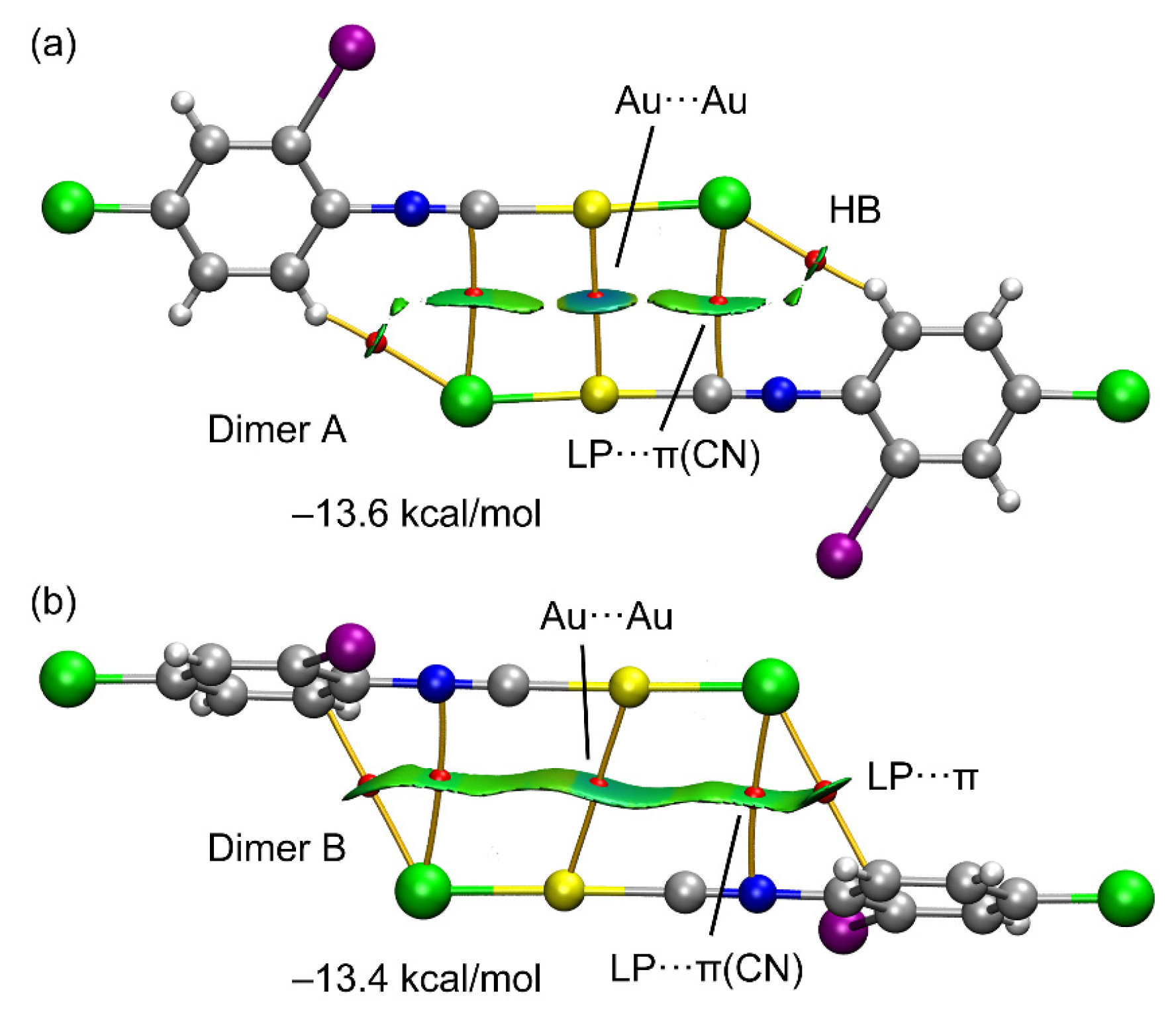
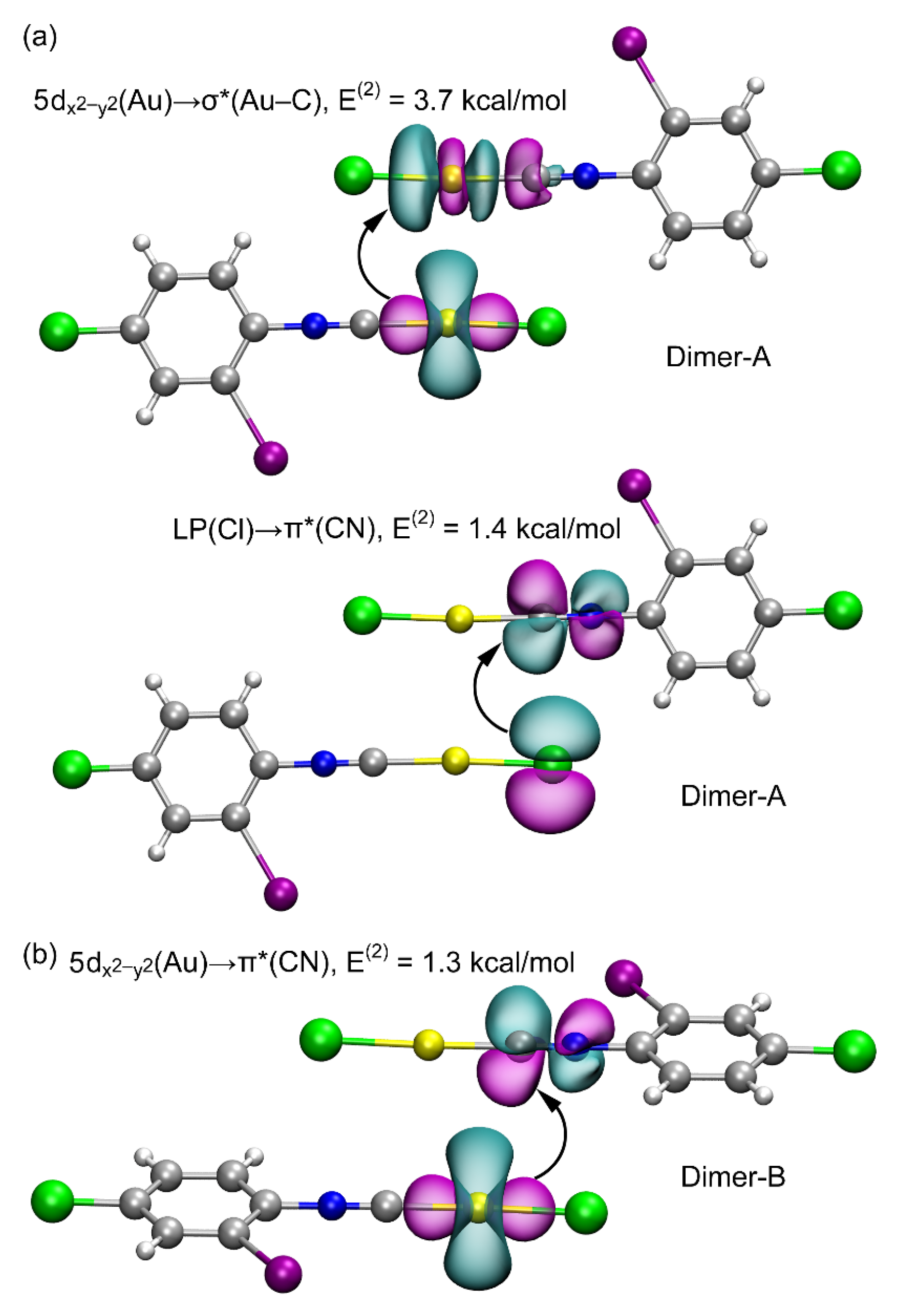
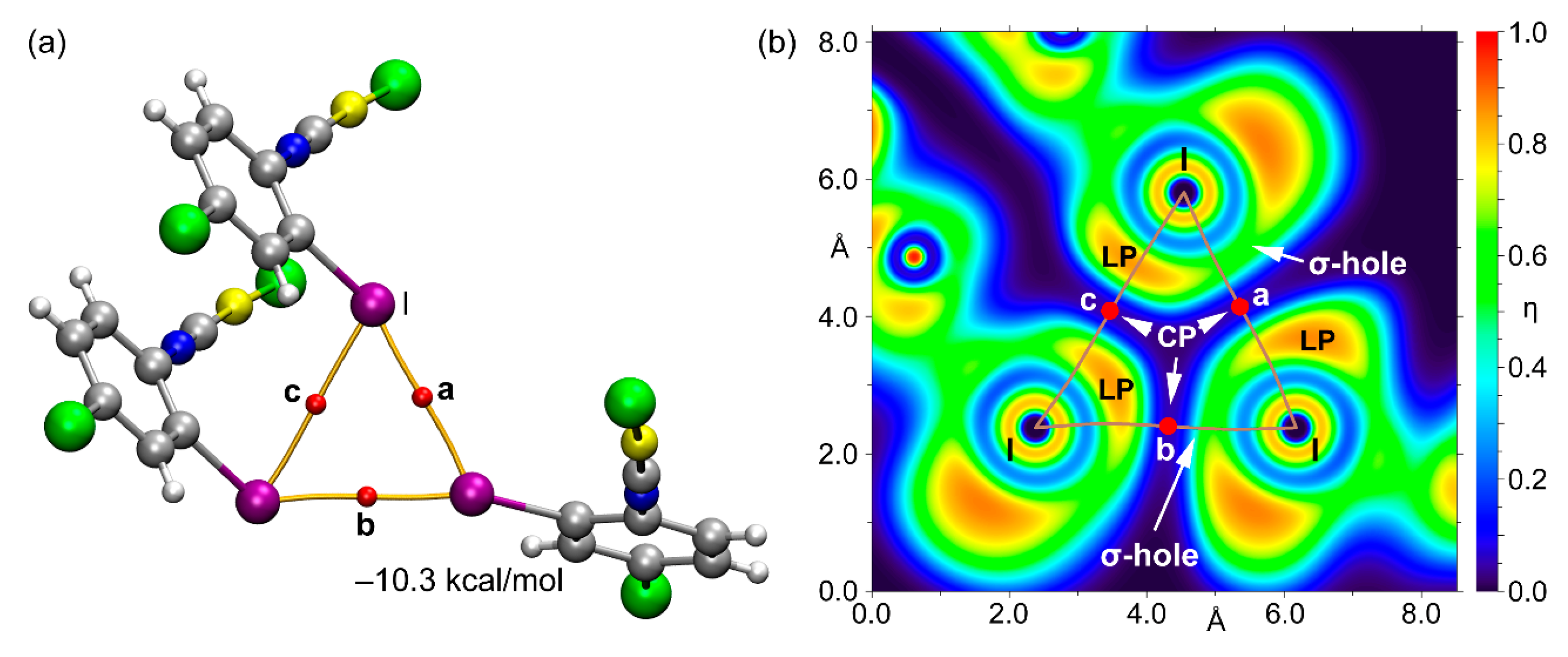
3.2. Theoretical Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shukla, R.; Chopra, D. Chalcogen and pnictogen bonds: Insights and relevance. Curr. Sci. 2021, 120, 1848–1853. [Google Scholar] [CrossRef]
- Cornaton, Y.; Djukic, J.-P. Noncovalent Interactions in Organometallic Chemistry: From Cohesion to Reactivity, a New Chapter. Acc. Chem. Res. 2021, 54, 3828–3840. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Frontera, A. Not Only Hydrogen Bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef] [Green Version]
- Frontera, A.; Bauzá, A. On the Importance of σ–Hole Interactions in Crystal Structures. Crystals 2021, 11, 1205. [Google Scholar] [CrossRef]
- Miller, D.K.; Chernyshov, I.Y.; Torubaev, Y.V.; Rosokha, S.V. From weak to strong interactions: Structural and electron topology analysis of the continuum from the supramolecular chalcogen bonding to covalent bonds. Phys. Chem. Chem. Phys. 2022, 8251–8259. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.-J. Noncovalent interactions: A brief account of a long history. J. Phys. Org. Chem. 2022, 35, e4340. [Google Scholar] [CrossRef]
- Non-covalent Interactions in the Synthesis and Design of New Compounds; Maharramov, A.M.; Mahmudov, K.T.; Kopylovich, M.N.; Pombeiro, A.J.L. (Eds.) Wiley Online Library: Hoboken, NJ, USA, 2016. [Google Scholar]
- Loh, C.C.J. Exploiting non-covalent interactions in selective carbohydrate synthesis. Nat. Rev. Chem. 2021, 5, 792–815. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Gurbanov, A.V.; Guseinov, F.I.; Guedes da Silva, M.F.C. Noncovalent interactions in metal complex catalysis. Coord. Chem. Rev. 2019, 387, 32–46. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Kopylovich, M.N.; da Silva, M.F.C.G.; Pombeiro, A.J. Noncovalent Interactions in Catalysis; Royal Society of Chemistry: London, UK, 2019. [Google Scholar]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. The Bright Future of Unconventional σ/π-Hole Interactions. ChemPhysChem 2015, 16, 2496–2517. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Electrostatics and Polarization in σ- and π-Hole Noncovalent Interactions: An Overview. ChemPhysChem 2020, 21, 579–588. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The σ-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef] [PubMed]
- Seth, S.K.; Bauza, A.; Frontera, A. Quantitative analysis of weak non-covalent σ-hole and π-hole interactions. Monogr. Supramol. Chem. 2019, 26, 285–333. [Google Scholar]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Noncovalent bonds through sigma and Pi-hole located on the same molecule. guiding principles and comparisons. Molecules 2021, 26, 1740. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, W.; Jin, W.J. σ-Hole Bond vs π-Hole Bond: A Comparison Based on Halogen Bond. Chem. Rev. 2016, 116, 5072–5104. [Google Scholar] [CrossRef] [PubMed]
- Echeverría, J. Intermolecular Carbonyl···Carbonyl Interactions in Transition-Metal Complexes. Inorg. Chem. 2018, 57, 5429–5437. [Google Scholar] [CrossRef]
- Doppert, M.T.; van Overeem, H.; Mooibroek, T.J. Intermolecular π-hole/n→π* interactions with carbon monoxide ligands in crystal structures. Chem. Comm. 2018, 54, 12049–12052. [Google Scholar] [CrossRef]
- Ruigrok van der Werve, A.; van Dijk, Y.R.; Mooibroek, T.J. π-Hole/n→π* interactions with acetonitrile in crystal structures. Chem. Commun. 2018, 54, 10742–10745. [Google Scholar] [CrossRef]
- Toikka, Y.N.; Mikherdov, A.S.; Ivanov, D.M.; Mooibroek, T.J.; Bokach, N.A.; Kukushkin, V.Y. Cyanamides as π-Hole Donor Components of Structure-Directing (Cyanamide)···Arene Noncovalent Interactions. Cryst. Growth Des. 2020, 20, 4783–4793. [Google Scholar] [CrossRef]
- Vatsadze, S.Z.; Loginova, Y.D.; dos Passos Gomes, G.; Alabugin, I.V. Stereoelectronic Chameleons: The Donor–Acceptor Dichotomy of Functional Groups. Chem. Eur. J. 2017, 23, 3225–3245. [Google Scholar] [CrossRef]
- Gomes, G.d.P.; Loginova, Y.; Vatsadze, S.Z.; Alabugin, I.V. Isonitriles as Stereoelectronic Chameleons: The Donor–Acceptor Dichotomy in Radical Additions. J. Amer. Chem. Soc. 2018, 140, 14272–14288. [Google Scholar] [CrossRef] [Green Version]
- Kinzhalov, M.A.; Ivanov, D.M.; Melekhova, A.A.; Bokach, N.A.; Gomila, R.M.; Frontera, A.; Kukushkin, V.Y. Chameleonic metal-bound isocyanides: A π-donating CuI-center imparts nucleophilicity to the isocyanide carbon toward halogen bonding. Inorg. Chem. Front. 2022, 9, 1655–1665. [Google Scholar] [CrossRef]
- Katkova, S.A.; Mikherdov, A.S.; Kinzhalov, M.A.; Novikov, A.S.; Zolotarev, A.A.; Boyarskiy, V.P.; Kukushkin, V.Y. (Isocyano Group π-Hole)⋅⋅⋅[d -MII] Interactions of (Isocyanide)[MII] Complexes, in which Positively Charged Metal Centers (d8-M=Pt, Pd) Act as Nucleophiles. Chem. Eur. J. 2019, 25, 8590–8598. [Google Scholar] [CrossRef]
- Mikherdov, A.S.; Kinzhalov, M.A.; Novikov, A.S.; Boyarskiy, V.P.; Boyarskaya, I.A.; Avdontceva, M.S.; Kukushkin, V.Y. Ligation-Enhanced π-Hole···π Interactions Involving Isocyanides: Effect of π-Hole···π Noncovalent Bonding on Conformational Stabilization of Acyclic Diaminocarbene Ligands. Inorg. Chem. 2018, 57, 6722–6733. [Google Scholar] [CrossRef]
- Mikherdov, A.S.; Katkova, S.A.; Novikov, A.S.; Efremova, M.M.; Reutskaya, E.Y.; Kinzhalov, M.A. (Isocyano group)⋯lone pair interactions involving coordinated isocyanides: Experimental, theoretical and CSD studies. CrystEngComm 2020, 22, 1154–1159. [Google Scholar] [CrossRef]
- Schmidbaur, H.; Schier, A. Aurophilic interactions as a subject of current research: An up-date. Chem. Soc. Rev. 2012, 41, 370–412. [Google Scholar] [CrossRef] [PubMed]
- Schmidbaur, H.; Schier, A. A briefing on aurophilicity. Chem. Soc. Rev. 2008, 37, 1931–1951. [Google Scholar] [CrossRef]
- Tiekink, E.R.T. Supramolecular assembly of molecular gold(I) compounds: An evaluation of the competition and complementarity between aurophilic (Au⋯Au) and conventional hydrogen bonding interactions. Coord. Chem. Rev. 2014, 275, 130–153. [Google Scholar] [CrossRef]
- Mirzadeh, N.; Privér, S.H.; Blake, A.J.; Schmidbaur, H.; Bhargava, S.K. Innovative Molecular Design Strategies in Materials Science Following the Aurophilicity Concept. Chem. Rev. 2020, 120, 7551–7591. [Google Scholar] [CrossRef]
- Weber, D.; Gagné, M.R. Aurophilicity in Gold(I) Catalysis: For Better or Worse? In Homogeneous Gold Catalysis; Slaughter, L.M., Ed.; Springer International Publishing: Cham, Switherland, 2015; pp. 167–211. [Google Scholar]
- Min, H.; Xiao, G.; Liu, W.; Liang, Y. Copper-catalyzed synthesis of 2-aminobenzothiazoles from 2-iodophenyl isocyanides, potassium sulfide and amines. Org. Biomol. Chem. 2016, 14, 11088–11091. [Google Scholar] [CrossRef] [PubMed]
- Uson, R.; Laguna, A.; Laguna, M.; Briggs, D.A.; Murray, H.H.; Fackler, J.P., Jr. (Tetrahydrothiophene)Gold(I) or Gold(III) Complexes. Inorg. Synth. 1989, 26, 85–91. [Google Scholar]
- Sheldrick, G. SHELXT - Integrated space-group and crystal-structure determination. Acta Cryst. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SADABS-2008/1-Bruker AXS Area Detector Scaling and Absorption Correction; Bruker AXS: Madison, WI, USA, 2008. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theor. Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Yamada, S.; Rokusha, Y.; Kawano, R.; Fujisawa, K.; Tsutsumi, O. Mesogenic gold complexes showing aggregation-induced enhancement of phosphorescence in both crystalline and liquid-crystalline phases. Faraday Discuss. 2017, 196, 269–283. [Google Scholar] [CrossRef]
- Ruch, A.A.; Ellison, M.C.; Nguyen, J.K.; Kong, F.; Handa, S.; Nesterov, V.N.; Slaughter, L.M. Highly Sterically Encumbered Gold Acyclic Diaminocarbene Complexes: Overriding Electronic Control in Regiodivergent Gold Catalysis. Organometallics 2021, 40, 1416–1433. [Google Scholar] [CrossRef]
- Ecken, H.; Olmstead, M.M.; Noll, B.C.; Attar, S.; Schlyer, B.; Balch, A.L. Effects of anions on the solid state structures of linear gold(I) complexes of the type (o-xylyl isocyanide)gold(I) (monoanion). J. Chem. Soc. Dalton Trans. 1998, 3715–3720. [Google Scholar] [CrossRef]
- Fujisawa, K.; Okuda, Y.; Izumi, Y.; Nagamatsu, A.; Rokusha, Y.; Sadaike, Y.; Tsutsumi, O. Reversible thermal-mode control of luminescence from liquid-crystalline gold(I) complexes. J. Mater. Chem. C 2014, 2, 3549–3555. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii of Metals in Covalent Compounds. J. Phys. Chem. 1966, 70, 3006–3007. [Google Scholar] [CrossRef]
- Alvarez, S. A cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. [Google Scholar] [CrossRef] [Green Version]
- Hashmi, A.S.K.; Hengst, T.; Lothschütz, C.; Rominger, F. New and Easily Accessible Nitrogen Acyclic Gold(I) Carbenes: Structure and Application in the Gold-Catalyzed Phenol Synthesis as well as the Hydration of Alkynes. Adv. Synth. Catal. 2010, 352, 1315–1337. [Google Scholar] [CrossRef]
- Hobbollahi, E.; List, M.; Redhammer, G.; Zabel, M.; Monkowius, U. Structural and photophysical characterization of gold(I) complexes bearing naphthyl chromophores. Inorg. Chem. Commun. 2016, 65, 24–27. [Google Scholar] [CrossRef]
- Katkova, S.A.; Mikherdov, A.S.; Sokolova, E.V.; Novikov, A.S.; Starova, G.L.; Kinzhalov, M.A. Intermolecular (Isocyano group)···PtII interactions involving coordinated isocyanides in cyclometalated PtII complexes. J. Mol. Struct. 2022, 1253, 132230. [Google Scholar] [CrossRef]
- Mikherdov, A.S.; Jin, M.; Ito, H. Exploring Au(i) involving halogen bonding with N-heterocyclic carbene Au(i) aryl complexes in crystalline media. Chem. Sci. 2023, 14, 4485–4494. [Google Scholar] [CrossRef] [PubMed]
- Aliyarova, I.S.; Tupikina, E.Y.; Ivanov, D.M.; Kukushkin, V.Y. Metal-Involving Halogen Bonding Including Gold(I) as a Nucleophilic Partner. The Case of Isomorphic Dichloroaurate(I)·Halomethane Cocrystals. Inorg. Chem. 2022, 61, 2558–2567. [Google Scholar] [CrossRef] [PubMed]
- Aliyarova, I.S.; Tupikina, E.Y.; Soldatova, N.S.; Ivanov, D.M.; Postnikov, P.S.; Yusubov, M.; Kukushkin, V.Y. Halogen Bonding Involving Gold Nucleophiles in Different Oxidation States. Inorg. Chem. 2022, 61, 15398–15407. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Saeed, R.R.A.; Shehata, M.N.I.; Ahmed, M.N.; Shawky, A.M.; Khowdiary, M.M.; Elkaeed, E.B.; Soliman, M.E.S.; Moussa, N.A.M. Type I-IV Halogen⋯Halogen Interactions: A Comparative Theoretical Study in Halobenzene⋯Halobenzene Homodimers. Int. J. Mol. Sci. 2022, 23, 3114. [Google Scholar] [CrossRef]
- Selvakumar, K.; Singh, H.B. Adaptive responses of sterically confined intramolecular chalcogen bonds. Chem. Sci. 2018, 9, 7027–7042. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||||
| φ is a torsion angle ClAuAuC | ||||||||
| CSD Code | d1 | d2 | d3 | d4 | ∠1 | ∠2 | ∠3 | φ |
| EBEZAP | 2.258(4) | 1.929(10) | 3.3159(8) | 3.373(11) | 94.0(3) | 86.7(2) | 91.6(4) | 0.1(3) |
| EQOGOJ | 2.2518(11) 2.2577(11) 2.2530(13) 2.2593(14) | 1.923(5) 1.919(4) 1.918(5) 1.935(5) | 3.2814(6) 3.2903(7) | 3.414(5) 3.436(5) 3.429(5) 3.448(4) | 87.34(13) 87.77(13) 87.63(13) 87.37(12) | 79.13(8) 79.27(8) 78.94(9) 79.29(9) | 97.32(16) 97.71(15) 97.36(15) 97.45(16) | 7.57(15) 8.88(14) 8.37(13) 9.57(14) |
| QIZTEA | 2.2564(15) 2.2584(14) | 1.946(6) 1.942(6) | 3.3092(6) | 3.433(6) | 90.64(16) 89.91(16) | 81.92(10) 82.57(10) | 95.24(19) 94.5(2) | 3.20(17) 2.88(17) |
| KEJRUQ | 2.255(2) 2.258(2) | 1.957(9) 1.939(9) | 3.3275(7) 3.2601(7) | 3.386(8) 3.410(8) | 101.5(2) 92.0(2) | 93.51(16) 82.92(18) | 83.8(2) 93.4(3) | 1.53(19) 1.12(18) |
| 1 | 2.2639(11) | 1.926(6) | 3.3050(8) | 3.428(6) | 92.53(16) | 83.77(10) | 93.15(19) | 0.90(17) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smirnov, A.S.; Kinzhalov, M.A.; Gomila, R.M.; Frontera, A.; Bokach, N.A.; Kukushkin, V.Y. Isocyanide π-Hole Interactions Supported by Aurophilic Forces. Crystals 2023, 13, 1177. https://doi.org/10.3390/cryst13081177
Smirnov AS, Kinzhalov MA, Gomila RM, Frontera A, Bokach NA, Kukushkin VY. Isocyanide π-Hole Interactions Supported by Aurophilic Forces. Crystals. 2023; 13(8):1177. https://doi.org/10.3390/cryst13081177
Chicago/Turabian StyleSmirnov, Andrey S., Mikhail A. Kinzhalov, Rosa M. Gomila, Antonio Frontera, Nadezhda A. Bokach, and Vadim Yu. Kukushkin. 2023. "Isocyanide π-Hole Interactions Supported by Aurophilic Forces" Crystals 13, no. 8: 1177. https://doi.org/10.3390/cryst13081177
APA StyleSmirnov, A. S., Kinzhalov, M. A., Gomila, R. M., Frontera, A., Bokach, N. A., & Kukushkin, V. Y. (2023). Isocyanide π-Hole Interactions Supported by Aurophilic Forces. Crystals, 13(8), 1177. https://doi.org/10.3390/cryst13081177