
Mn(III)–Salen Complexes with Metallophilic Interactions


Abstract
:1. Introduction
2. Experimental Section
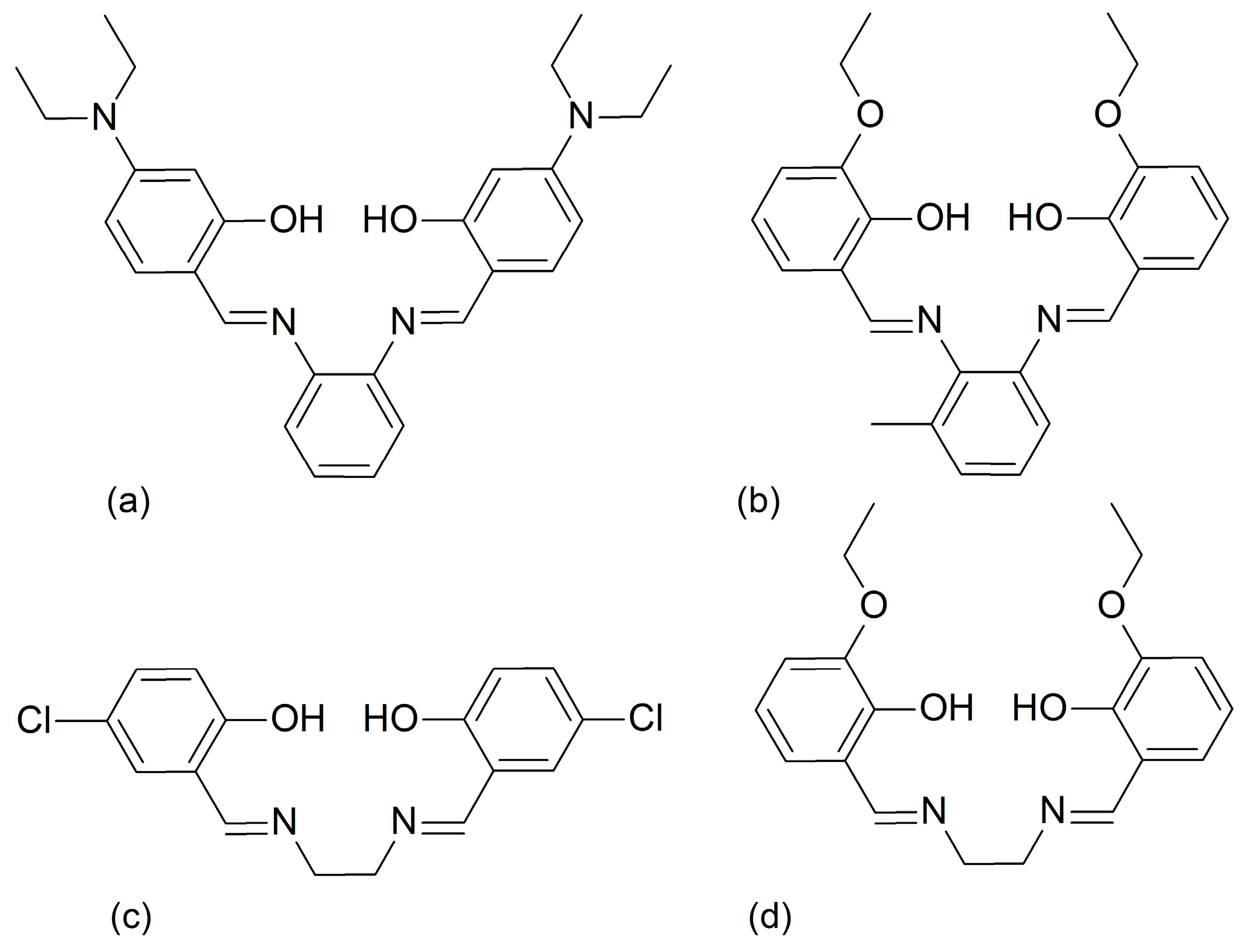
2.1. Synthesis
2.2. Measurements
2.3. Crystallography
2.4. Theoretical Calcualtions
3. Results
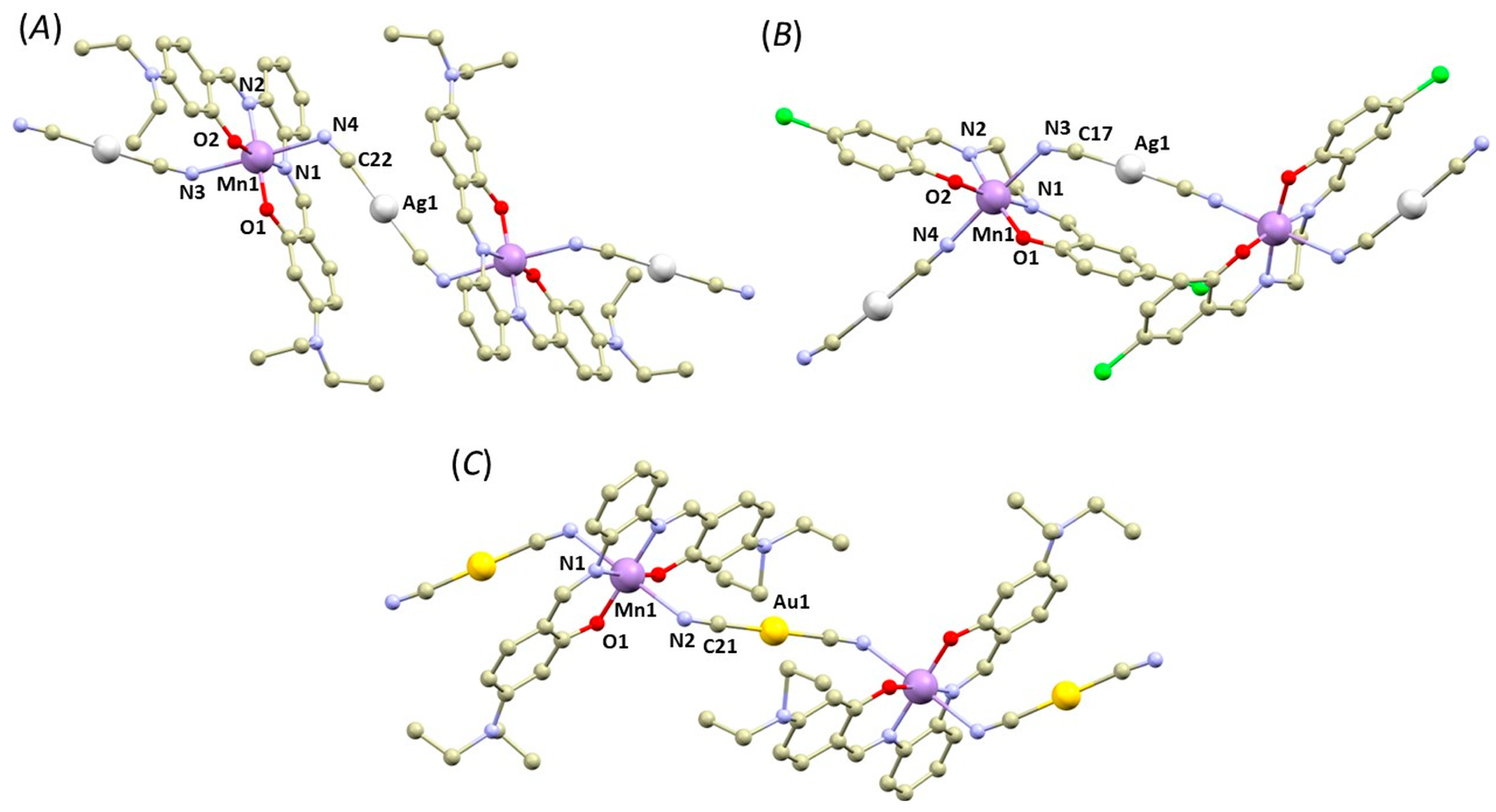
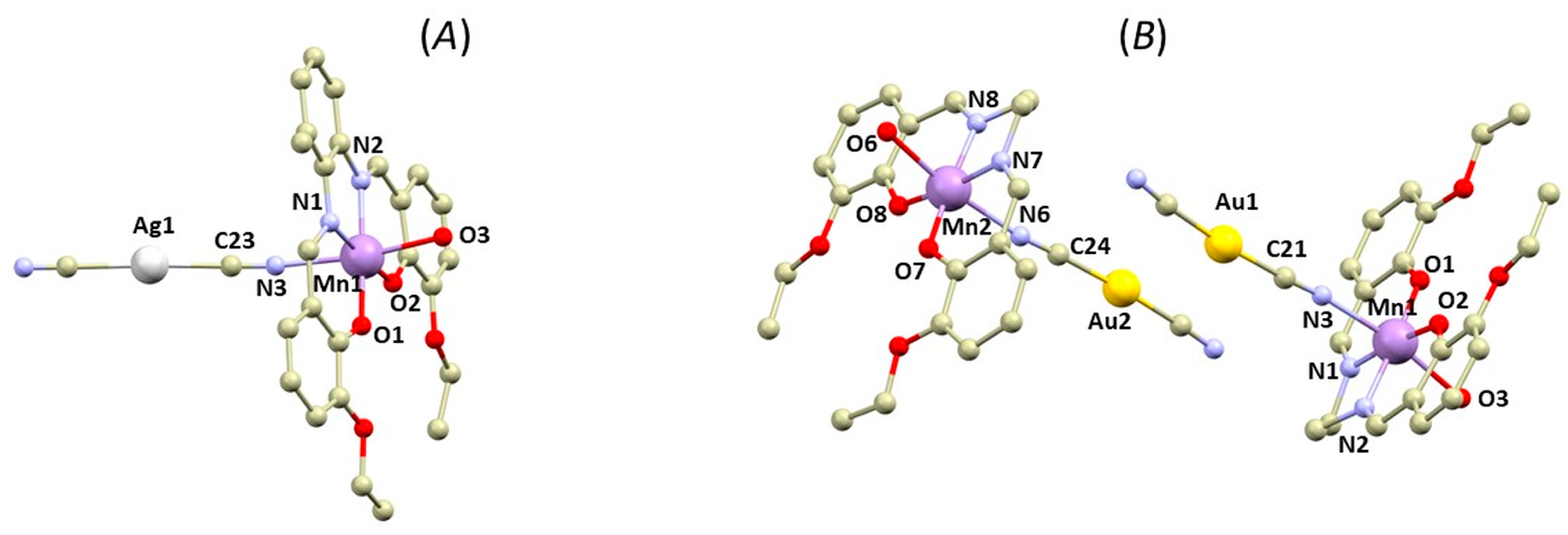
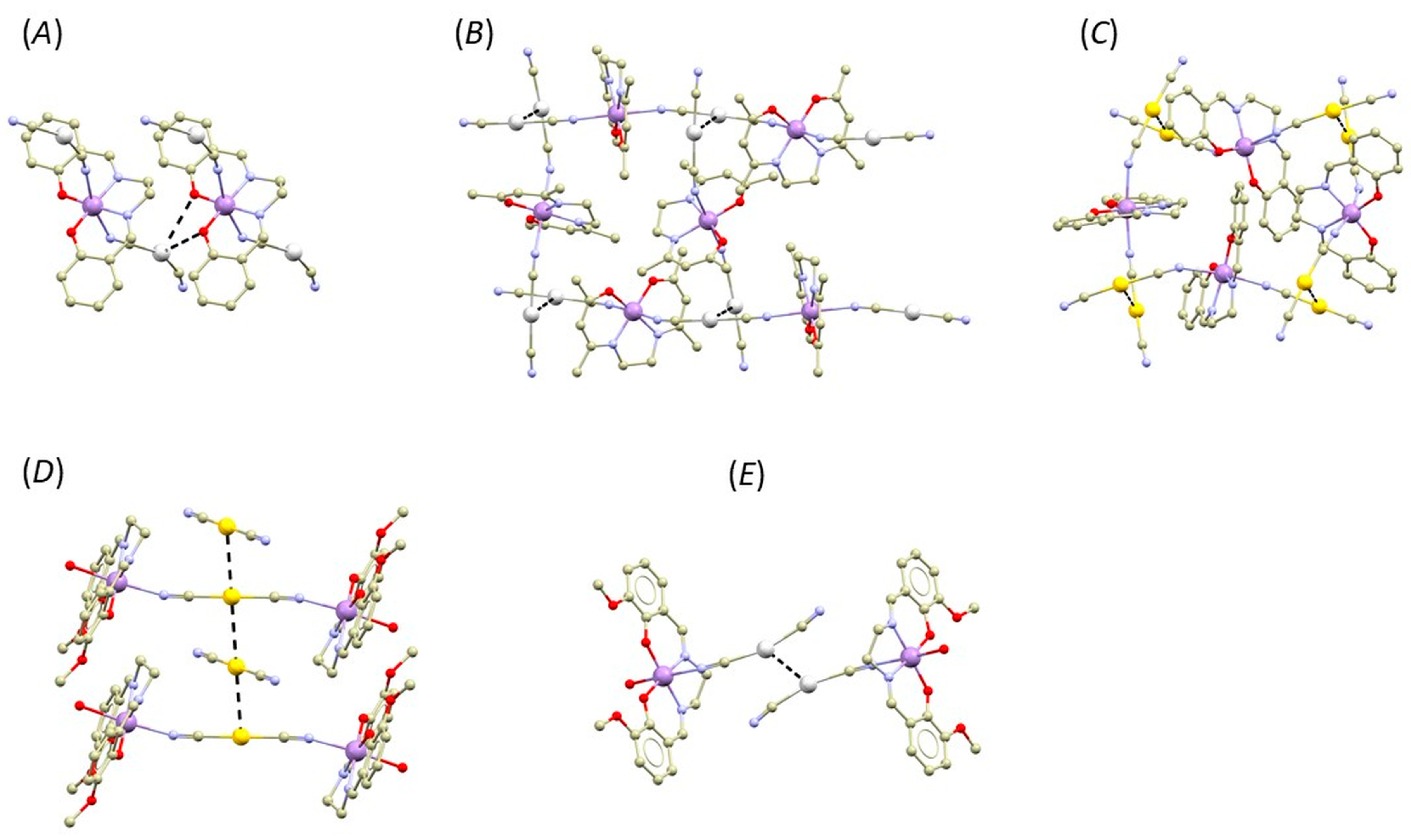
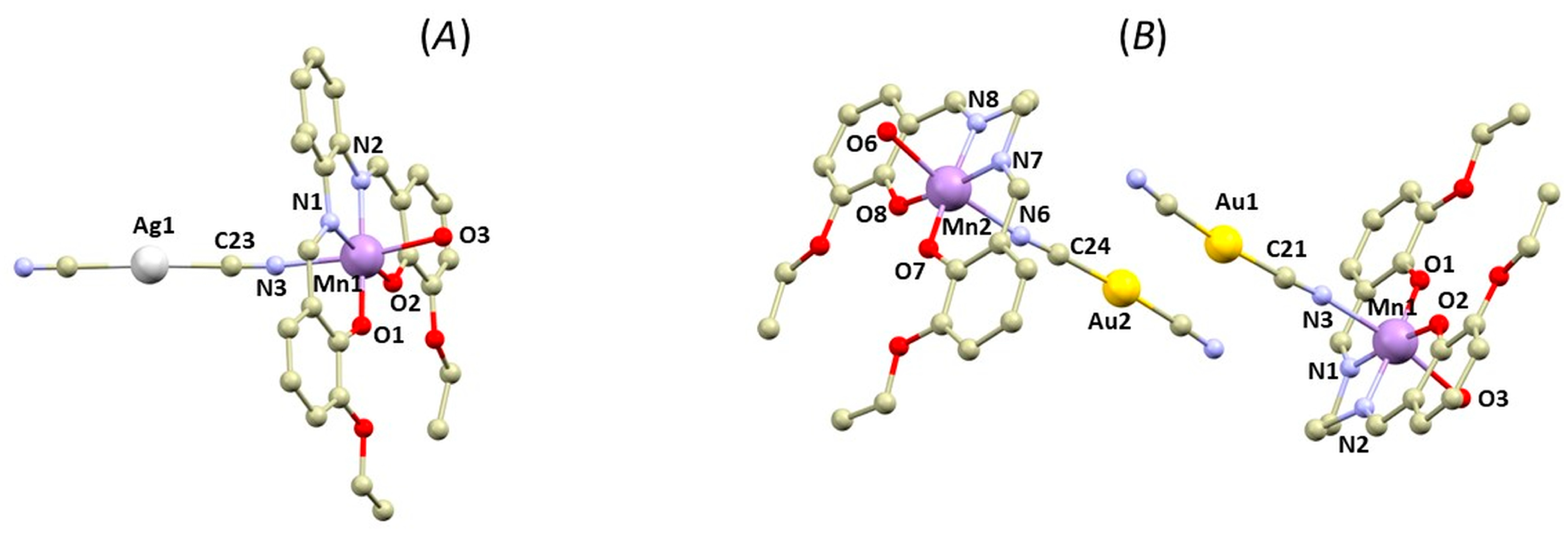
3.1. Structural Overview
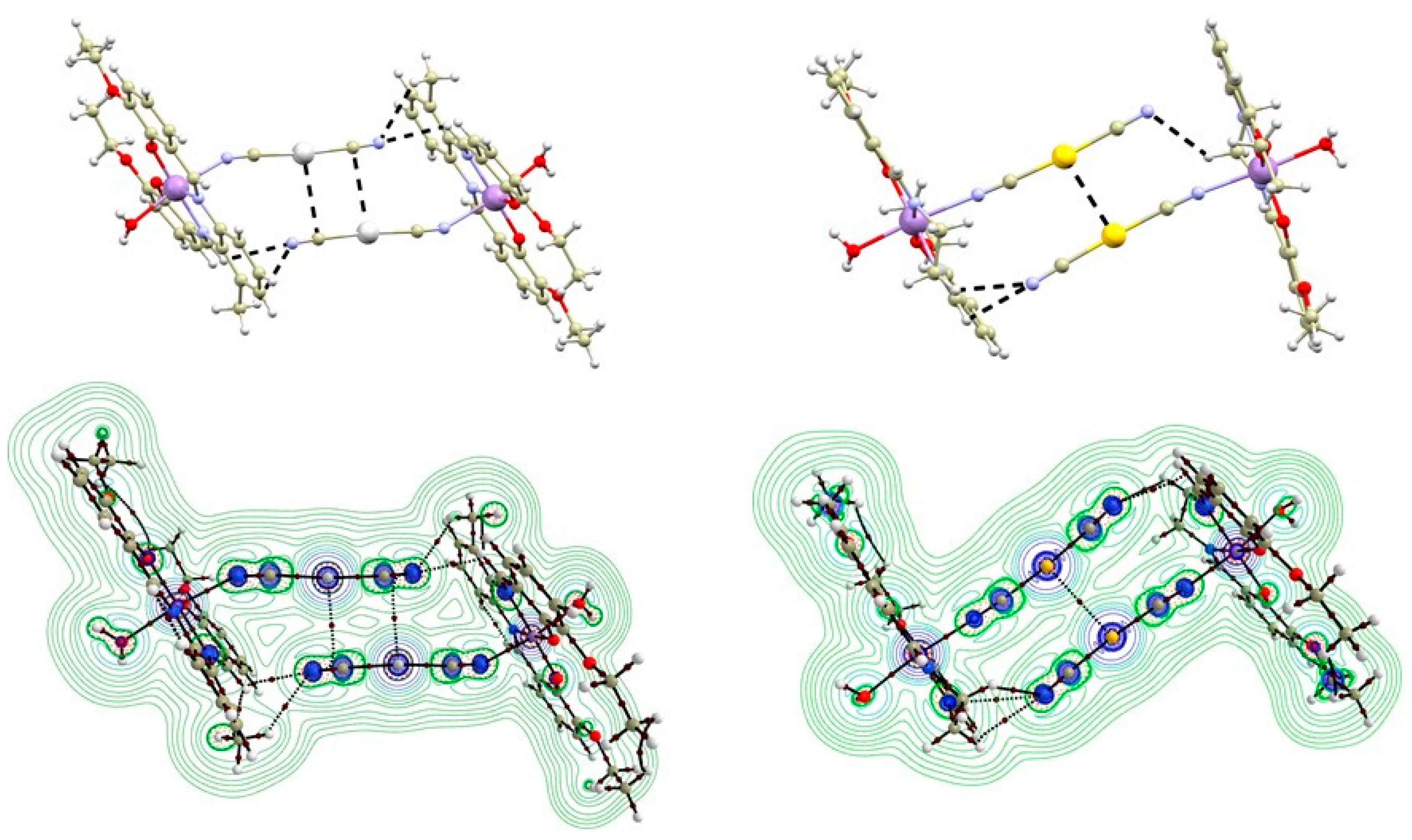
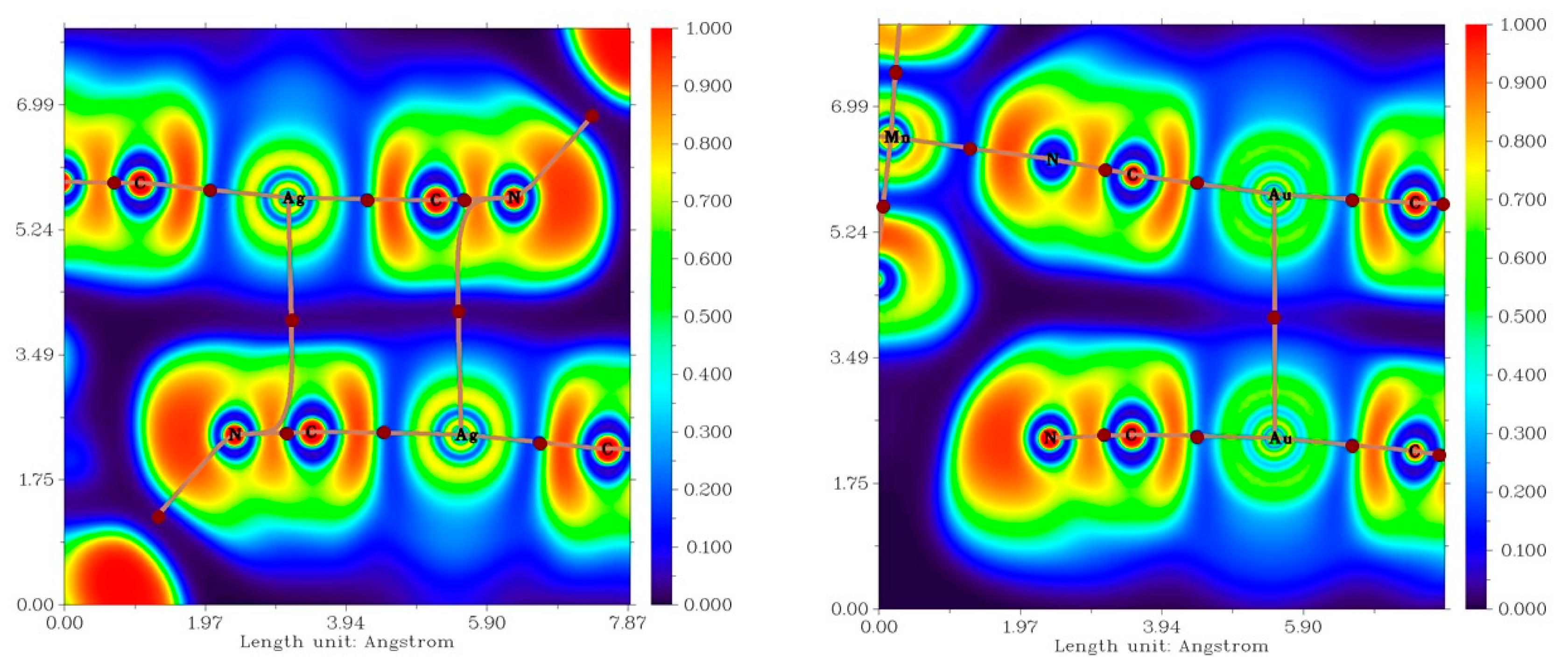
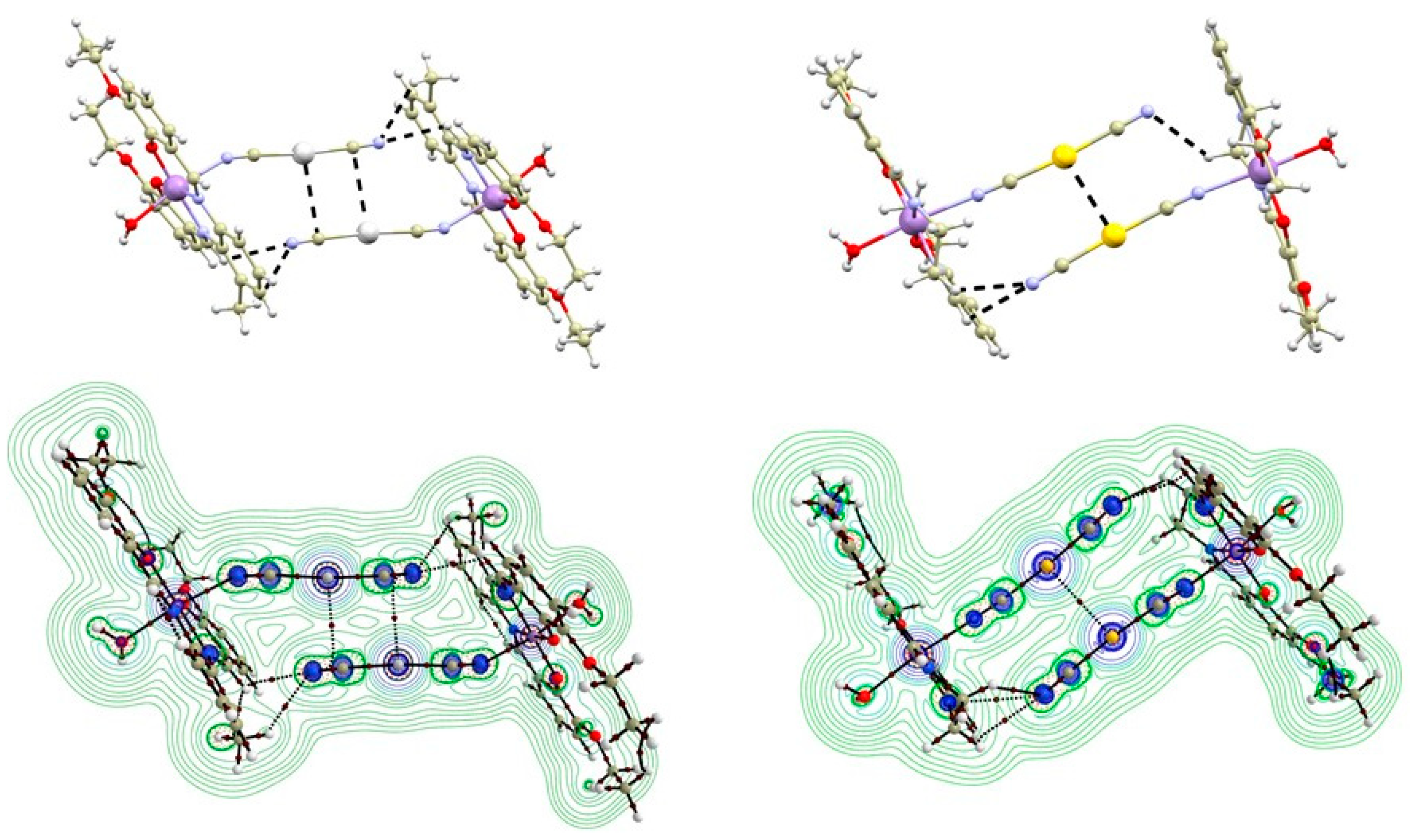
3.2. QT–AIM Analysis of Non-Covalent Interactions
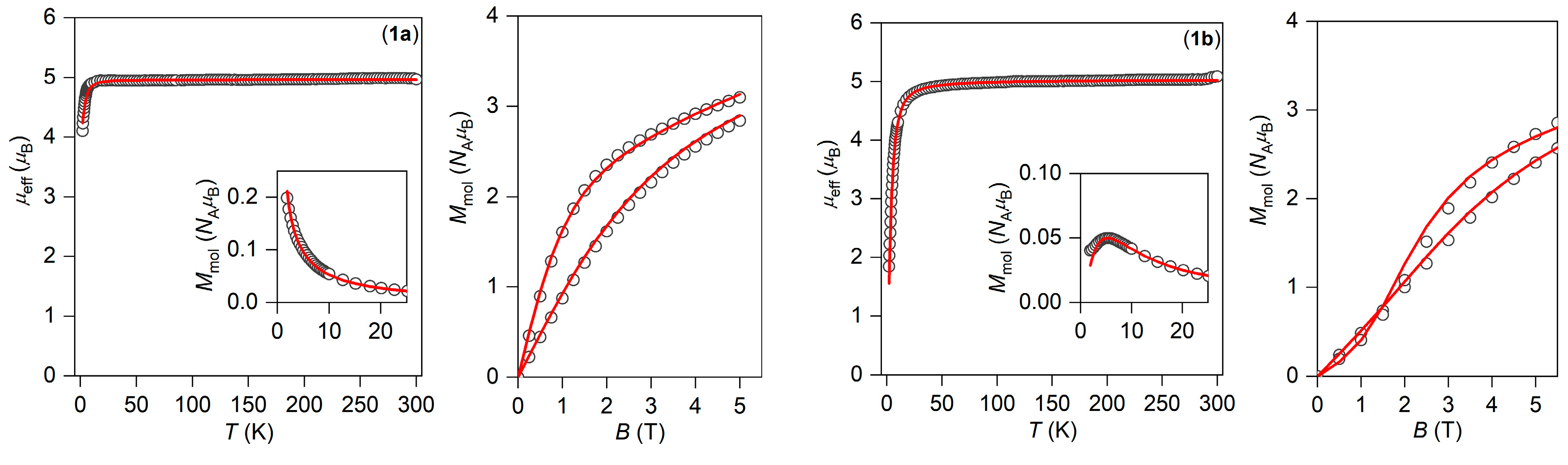
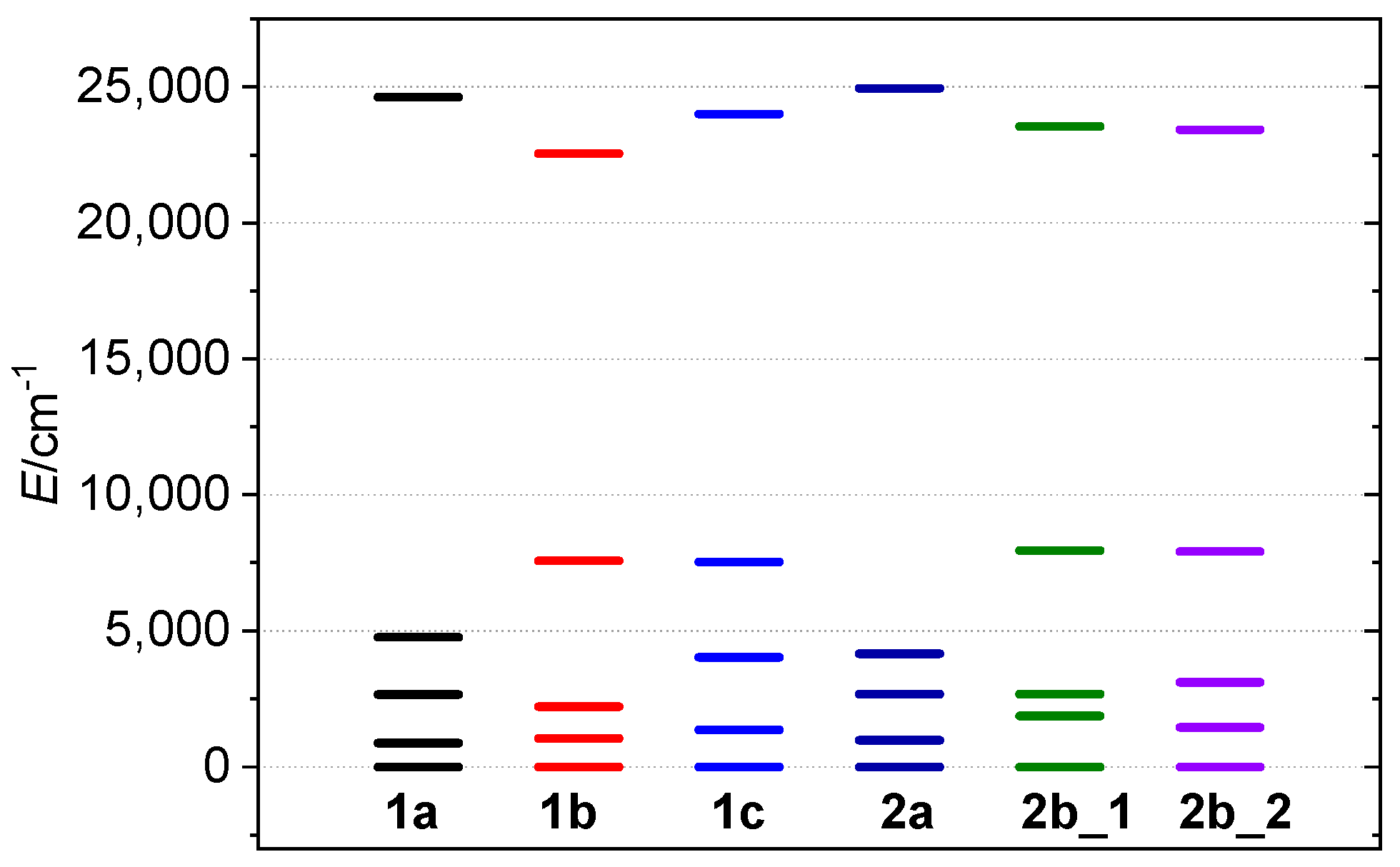
3.3. Experimental and Theoretical Investigation of Magnetic Properties
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gao, S. Molecular Nanomagnets and Related Phenomena. In Structure and Bonding; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Kachi-Terajima, C.; Ishii, R.; Tojo, Y.; Fukuda, M.; Kitagawa, Y.; Asaoka, M.; Miyasaka, H. Ferromagnetic Exchange Coupling in a Family of MnIII Salen-Type Schiff-Base Out-of-Plane Dimers. J. Phys. Chem. C 2017, 121, 12454–12468. [Google Scholar] [CrossRef]
- Bogani, L.; Vindigni, A.; Sessoli, R.; Gatteschi, D. Single chain magnets: Where to from here? J. Mater. Chem. 2008, 18, 4750–4758. [Google Scholar] [CrossRef]
- Ferbinteanu, M.; Miyasaka, H.; Wernsdorfer, W.; Nakata, K.; Sugiura, K.; Yamashita, M.; Coulon, C.; Clérac, R. Single-Chain Magnet (NEt4)[Mn2(5-MeOsalen)2Fe(CN)6] Made of MnIII−FeIII−MnIII Trinuclear Single-Molecule Magnet with an ST = 9/2 Spin Ground State. J. Am. Chem. Soc. 2005, 127, 3090–3099. [Google Scholar] [CrossRef]
- Nemec, I.; Herchel, R.; Šilha, T.; Trávníček, Z. Towards a better understanding of magnetic exchange mediated by hydrogen bonds in Mn(III)/Fe(III) salen-type supramolecular dimers. Dalton Trans. 2014, 43, 15602–15616. [Google Scholar] [CrossRef] [Green Version]
- Nemec, I.; Šilha, T.; Herchel, R.; Trávníček, Z. Investigation of Magnetic Exchange Pathways in Heterotrinuclear Manganese(III) Schiff Base Complexes Involving Tetrathiocyanidoplatinate(II) Bridges. Eur. J. Inorg. Chem. 2013, 2013, 5781–5789. [Google Scholar] [CrossRef]
- Šilha, T.; Nemec, I.; Herchel, R.; Trávníček, Z. Structural and Magnetic Characterizations of the First Manganese(III) Schiff Base Complexes Involving Hexathiocyanidoplatinate(IV) Bridges. CrystEngComm 2013, 15, 5351–5358. [Google Scholar] [CrossRef]
- Nemec, I.; Herchel, R.; Trávníček, Z. Pentacoordinate and Hexacoordinate Mn(III) Complexes of Tetradentate Schiff-Base Ligands Containing Tetracyanidoplatinate(II) Bridges and Revealing Uniaxial Magnetic Anisotropy. Molecules 2016, 21, 1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkorta, I.; Elguero, J.; Frontera, A. Not Only Hydrogen Bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef] [Green Version]
- Raju, S.; Singh, H.B.; Butcher, R.J. Metallophilic Interactions: Observations of the Shortest Metallophilicinteractions between Closed Shell (d10⋯d10, d10⋯d8, d8⋯d8) Metal Ions [M⋯M′ M = Hg(ii) and Pd(ii) and M′ = Cu(i), Ag(i), Au(i), and Pd(ii)]. Dalton Trans. 2020, 49, 9099–9117. [Google Scholar] [CrossRef]
- Zheng, Q.; Borsley, S.; Nichol, G.S.; Duarte, F.; Cockroft, S.L. The Energetic Significance of Metallophilic Interactions. Angew. Chem. Int. Ed. 2019, 58, 12617–12623. [Google Scholar] [CrossRef]
- Panja, A.; Shaikh, N.; Vojtíšek, P.; Gaoc, S.; Banerjee, P. Synthesis, crystal structures and magnetic properties of 1D polymeric [MnIII(salen)N3] and [MnIII(salen)Ag(CN)2] complexes. New J. Chem. 2002, 26, 1025–1028. [Google Scholar] [CrossRef]
- Kosone, Y.; Suzuki, Y.; Kanadani, C.; Saito, T.; Kitazawa, T. Structural isomers of {MnII(L)2[AgI(CN)2]2} (L = 3-methylpyridine or 4-methylpyridine), bilayer structure with binuclear argentophilic interaction and interpenetrated structure with 1D chain argentophilic interaction; synthesis, crystal structure, and magnetic properties. Bull. Chem. Soc. Jpn. 2009, 82, 347–351. [Google Scholar]
- Feng, Y.; Guo, Y.; Yang, Y.O.; Liu, Z.; Liao, D.; Cheng, P.; Yan, S.; Jiang, Z. 2D warp-and-woof interwoven networks constructed by helical chains with different chirality. Chem. Commun. 2007, 35, 3643–3645. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, H.; Tian, L.; Jiang, J.; Ni, Z.H. Rational design of cyanide-bridged heterometallic M(I)–Mn(II) (M = Ag, Au) one-dimensional chain complexes: Synthesis, crystal structures and magnetic properties. CrystEngComm 2009, 11, 2447–2451. [Google Scholar] [CrossRef]
- Guo, Y.; Ma, Y.; Zhou, N.; Liu, Z.Q.; Wang, Q.L.; Yan, S.P.; Liao, D.Z. Three dicyanidometallate(I)-based complexes incorporating hydrogenbonding, π–π packing and d10d10 interactions with auxiliary 2,2′-bipyridyl like ligands. Z. Anorg. Allg. Chem. 2010, 636, 865–871. [Google Scholar] [CrossRef]
- Nastase, S.; Tuna, F.; Maxim, C.; Muryn, C.A.; Avarvari, N.; Winpenny, R.E.P.; Andruh, M. Supramolecular dimers and chains resulting from second coordination sphere interactions. Cryst. Growth Des. 2007, 7, 1825–1831. [Google Scholar] [CrossRef]
- Jayaseeli, A.M.I.; Ramdass, A.; Rajagopal, S. Selective H2O2 oxidation of organic sulfides to sulfoxides catalyzed by cobalt (III)–salen ionw. Polyhedron 2015, 100, 59–66. [Google Scholar] [CrossRef]
- Gravert, D.J.; Griffin, J.H. Steric and electronic effects, enantiospecificity, and reactive orientation in DNA binding/cleaving by substituted derivatives of [SalenMnIII]+. Inorg. Chem. 1996, 35, 4837–4847. [Google Scholar] [CrossRef]
- CrysAlis CCD and CrysAlis RED, version 1.171.33.52; Oxford Diffraction Ltd.: Oxford, UK, 2009.
- CrysAlisPro, version 1.171.40.82a; Rigaku Oxford Diffraction: Oxford, UK, 2020.
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. In Acta Crystallographica Section A Foundations and Advances; International Union of Crystallography (IUCr): Chester, UK, 2015; Volume 71, pp. 3–8. [Google Scholar]
- Ruiz, E.; Cano, J.; Alvarez, S.; Alemany, P. Broken symmetry approach to calculation of exchange coupling constants for homobinuclear and heterobinuclear transition metal complexes. J. Comput. Chem. 1999, 20, 1391–1400. [Google Scholar] [CrossRef]
- Soda, T.; Kitagawa, Y.; Onishi, T.; Takano, Y.; Shigeta, Y.; Nagao, H.; Yoshioka, Y.; Yamaguchi, K. Ab initio computations of effective exchange integrals for H-H, H-He-H and Mn2O2 complex: Comparison of broken-symmetry approaches. Chem. Phys. Lett. 2000, 319, 223–230. [Google Scholar] [CrossRef]
- Todd, A.; Keith, T.K. AIMAll, version 19.10.12; Gristmill Software: Overland Park, KS, USA, 2019.
- Pyykkö, P. Strong Closed-Shell Interactions. Rev. Inorg. Chem. 1997, 97, 597–636. [Google Scholar]
- Schmidbaur, H.; Schier, A. Argentophilic interactions. Angew. Chem. Int. Ed. 2015, 54, 746–784. [Google Scholar] [CrossRef]
- Guevara-Vela, J.M.; Hess, K.; Rocha-Rinza, T.; Martín Pendás, Á.; Flores-Álamo, M.; Moreno-Alcántar, G. Stronger-Together: The Cooperativity of Aurophilic Interactions. Chem. Comm. 2022, 58, 1398–1401. [Google Scholar] [CrossRef]
- Eryazici, I.; Moorefield, C.N.; Newkome, G.R. Square-planar Pd(II), Pt(II), and Au(III) terpyridine complexes: Their syntheses, physical properties, supramolecular constructs, and biomedical activities. Chem. Rev. 2008, 108, 1834–1895. [Google Scholar] [CrossRef]
- Shedrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Frontera, A.; Bauzá, A. Regium-π bonds: An Unexplored Link between Noble Metal Nanoparticles and Aromatic Surfaces. Chem. Eur. J. 2018, 24, 7228–7234. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.W. Meaning and Functional Form of the Electron Localization Function. Acta Phys. Chim. Sin. 2011, 27, 2786–2792. [Google Scholar]
- Xiao, W.; Kiran, G.K.; Yoo, K.; Kim, J.; Xu, H. The Dual-Site Adsorption and High Redox Activity Enabled by Hybrid Organic-Inorganic Vanadyl Ethylene Glycolate for High-Rate and Long-Durability Lithium–Sulfur Batteries. Small 2023, 19, e2206750. [Google Scholar] [CrossRef]
- Xu, H.; Guan, D. Exceptional Anisotropic Noncovalent Interactions in Ultrathin Nanorods: The Terminal σ-Hole. ACS Appl. Mater. Interfaces 2022, 14, 51190–51199. [Google Scholar] [CrossRef]
- Lv, Z.; Xu, H.; Xu, W.; Peng, B.; Zhao, C.; Xie, M.; Lv, X.; Gao, Y.; Hu, K.; Fang, Y.; et al. Quasi-Topological Intercalation Mechanism of Bi0.67NbS2 Enabling 100 C Fast-Charging for Sodium-Ion Batteries. Adv. Energy Mater. 2023, 13, 2300790. [Google Scholar] [CrossRef]
- Singh, S.K.; Eng, J.; Atanasov, M.; Neese, F. Covalency and chemical bonding in transition metal complexes: An ab initio based ligand field perspective. Coord. Chem. Rev. 2017, 344, 2–25. [Google Scholar] [CrossRef]
- Boča, R. Theoretical Foundations of Molecular Magnetism. Elsevier: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Mingos, D.M.P.; Day, P.; Dahl, J.P. Molecular Electronic Structures of Transition Metal Complexes II; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Hellweg, A.; Hättig, C.; Höfener, S.; Klopper, W. Optimized accurate auxiliary basis sets for RI-MP2 and RI-CC2 calculations for the atoms Rb to Rn. Theor. Chem. Acc. 2007, 117, 587–597. [Google Scholar] [CrossRef]
- Izsák, R.; Neese, F. An overlap fitted chain of spheres exchange method. J. Chem. Phys. 2011, 135, 144105. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree–Fock and hybrid DFT calculations A ‘chain-of-spheres’ algorithm for the Hartree–Fock Exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From Weak to Strong Interactions: A Comprehensive Analysis of the Topological and Energetic Properties of the Electron Density Distribution Involving X–H⋯F–Y Systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Silviu, N.; Catalin, M.; Carine, D.; Jean-Pascal, S.; Marius, A. Synthesis and crystal structures of two new cyanido-bridged [MnIII5MoIV] and [MnIII2AuI] heterometallic complexes. Rev. Roum. Chem. 2013, 58, 355–363. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1a | 1b | 1c | 2a | 2b | |
|---|---|---|---|---|---|
| Mn–Nim | 1.974(5) | 2.000(3) | 1.9908(15) | 1.971(4) | 1.981 * |
| 1.976(5) | 1.999(3) | 1.9814(16) | 1.971(4) | 1.985 * | |
| Mn–NCN | 2.395(6) | 2.299(3) | 2.2605(19) | 2.467(6) | 2.273(2) 2.283(2) |
| 2.442(6) | 2.3894(18) | 2.467(6) | |||
| Mn–Oph | 1.866(4) | 1.881(2) | 1.8786(13) | 1.862(4) | 1.871 * |
| 1.872(4) | 1.882(2) | 1.8980(12) | 1.862(4) | 1.893 * | |
| Mn–Osolv | - | 2.249(2) | - | - | 2.2737(17) 2.2685(16) |
| Complex | gx | gy | gz | giso | D/cm−1 | E/D | J */cm−1 | zj/cm−1 |
|---|---|---|---|---|---|---|---|---|
| 1a exp | - | - | - | 2.027(2) | −3.0(1) | - | - | −0.03(1) |
| 1a calc | 1.982 | 1.997 | 1.998 | 1.992 | −2.84 | 0.01 | - | - |
| 1b exp | - | - | - | 2.056(3) | −4.3(2) | - | −0.58(2) | - |
| 1b calc | 1.980 | 1.997 | 1.997 | 1.991 | −3.13 | 0.01 | −0.46 | - |
| 1c exp | - | - | - | 2.037(5) | −3.2(2) | - | - | 0.09(1) |
| 1c calc | 1.979 | 1.997 | 1.998 | 1.991 | −3.09 | 0.03 | - | - |
| 2a exp ‡ | - | - | - | 1.943(5) | −2.81(7) | - | - | −0.20(1) |
| 2a calc | 1.982 | 1.997 | 1.998 | 1.992 | −2.80 | 0.00 | - | - |
| 2b exp | - | - | - | 1916(9) | −3.1(6) | - | −0.73(7) | - |
| 2b calc † Mn1 | 1.980 | 1.997 | 1.997 | 1.991 | −3.13 | 0.02 | −0.60 | - |
| Mn2 | 1.980 | 1.997 | 1.997 | 1.991 | −3.16 | 0.03 | −0.56 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šilha, T.; Herchel, R.; Nemec, I. Mn(III)–Salen Complexes with Metallophilic Interactions. Crystals 2023, 13, 1217. https://doi.org/10.3390/cryst13081217
Šilha T, Herchel R, Nemec I. Mn(III)–Salen Complexes with Metallophilic Interactions. Crystals. 2023; 13(8):1217. https://doi.org/10.3390/cryst13081217
Chicago/Turabian StyleŠilha, Tomáš, Radovan Herchel, and Ivan Nemec. 2023. "Mn(III)–Salen Complexes with Metallophilic Interactions" Crystals 13, no. 8: 1217. https://doi.org/10.3390/cryst13081217
APA StyleŠilha, T., Herchel, R., & Nemec, I. (2023). Mn(III)–Salen Complexes with Metallophilic Interactions. Crystals, 13(8), 1217. https://doi.org/10.3390/cryst13081217