
Crystal Structure Analysis, Stability, Phase Transformation and Selective Nucleation Mechanism of Fluralaner Polymorphs



 ,
, 
Abstract
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Preparation of Fluralaner Polymorphs
2.2.1. Preparation of Form I
2.2.2. Preparation of Form Ⅱ
2.2.3. Preparation of Form Ⅲ
2.3. Characterization
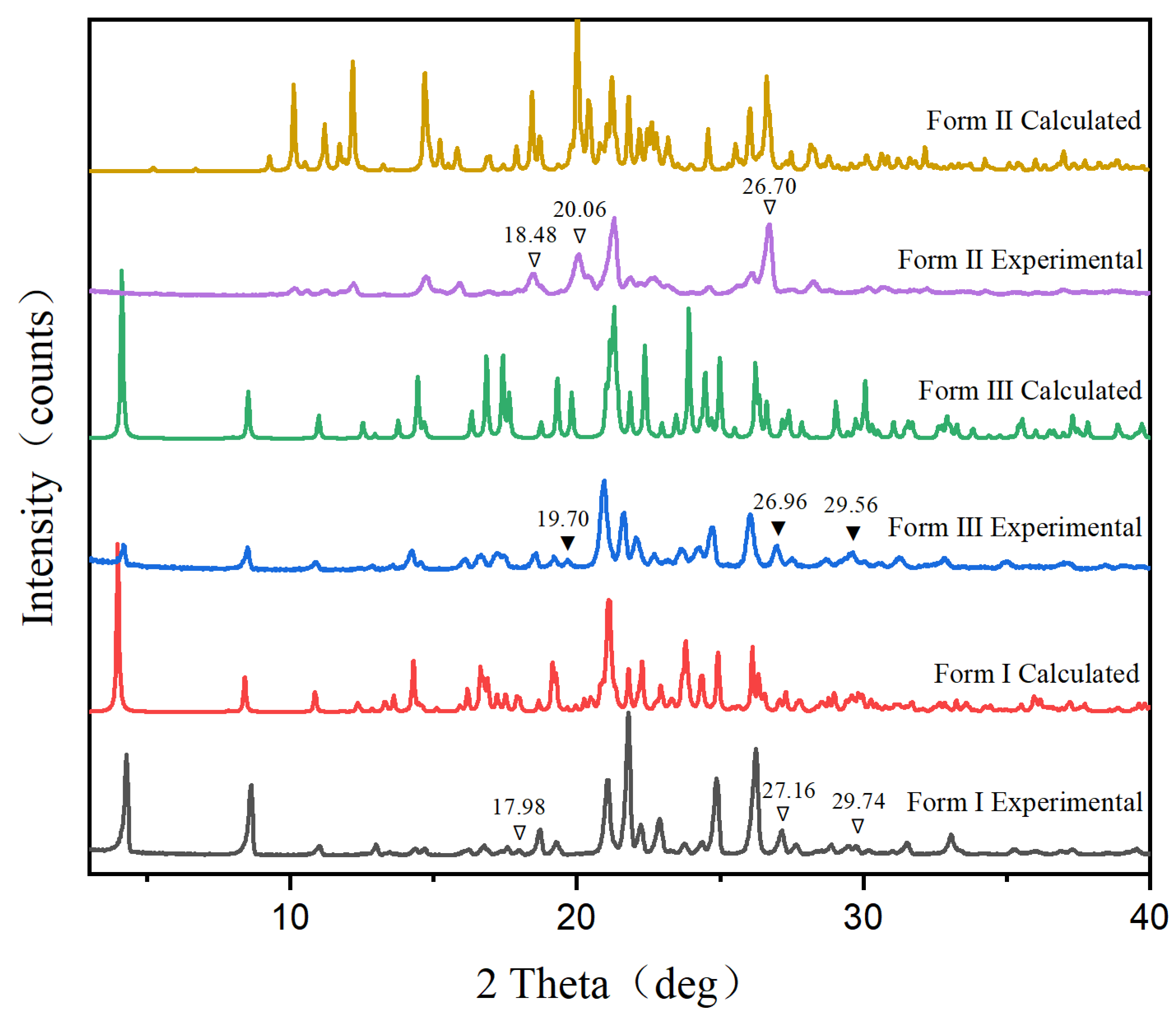
2.3.1. Powder X-ray Diffraction (PXRD)
2.3.2. Differential Scanning Calorimetry (DSC)
2.3.3. Thermogravimetric Analysis (TGA)
2.3.4. Morphology Analysis
2.3.5. Solid-State Nuclear Magnetic Resonance (SSNMR)
2.3.6. Fourier Transform Infrared (FTIR)
2.3.7. Single Crystal X-ray Diffraction (SCXRD)
2.4. Hirshfeld Surface Analysis
2.5. Solvent-Mediated Phase Transformation Experiment
2.6. Single Point Energy Calculations
2.7. Lattice Energy Calculation
2.8. Molecular Dynamics (MD) Simulations
2.9. Radial Distribution Function (RDF)
3. Results and Discussion
3.1. Characterization of Polymorphs
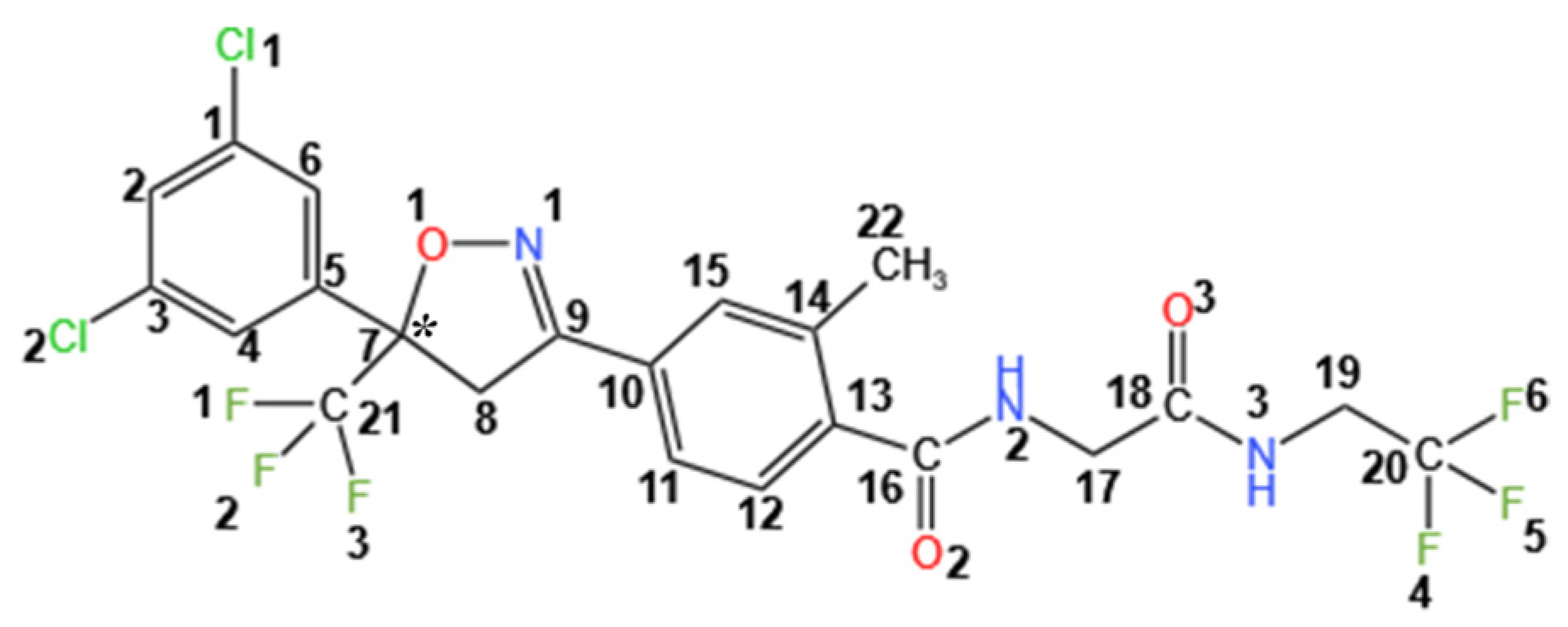
3.2. Molecular Conformations and Crystal Structure Analysis
3.2.1. Crystal Structure Analysis
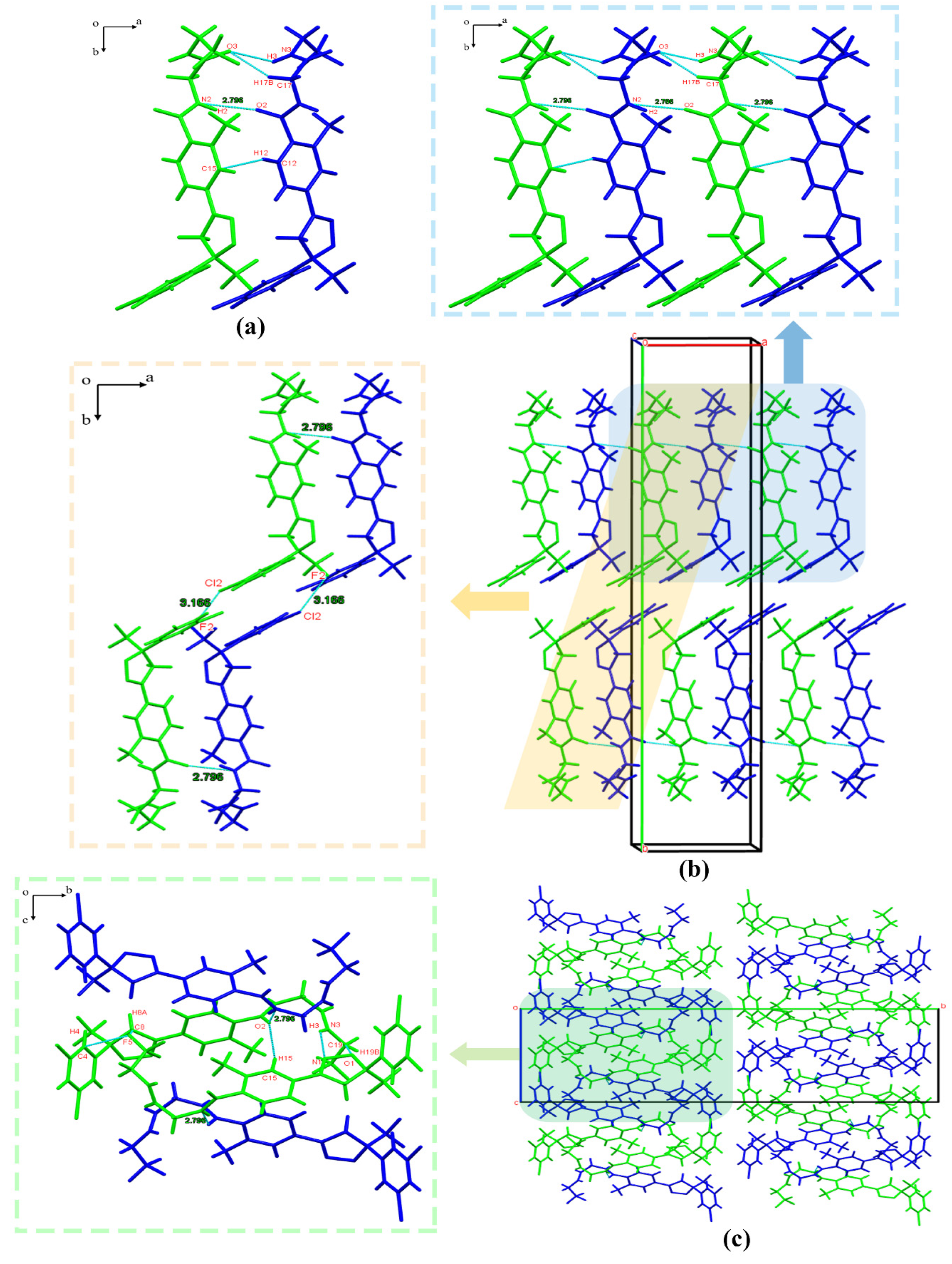
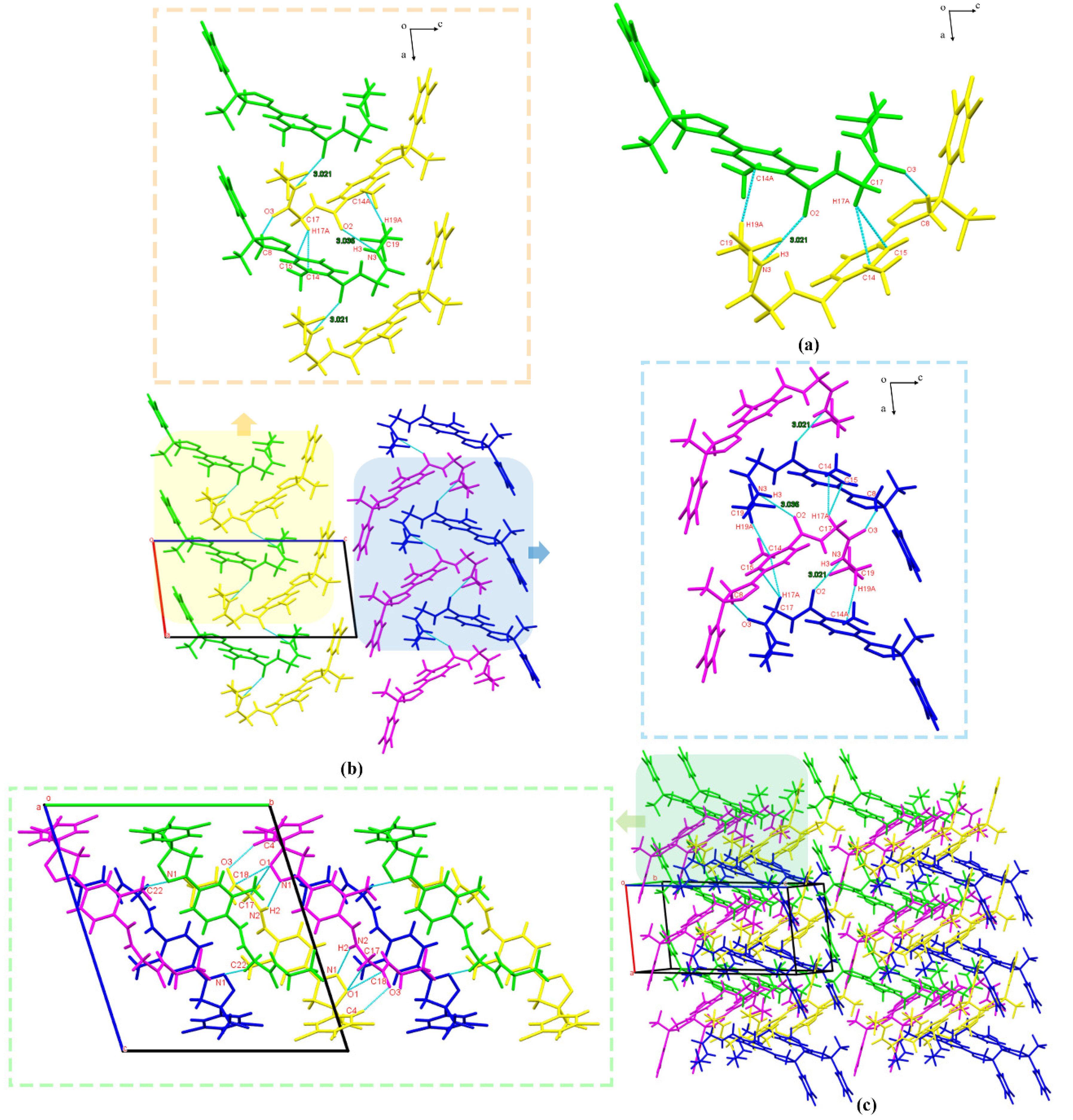
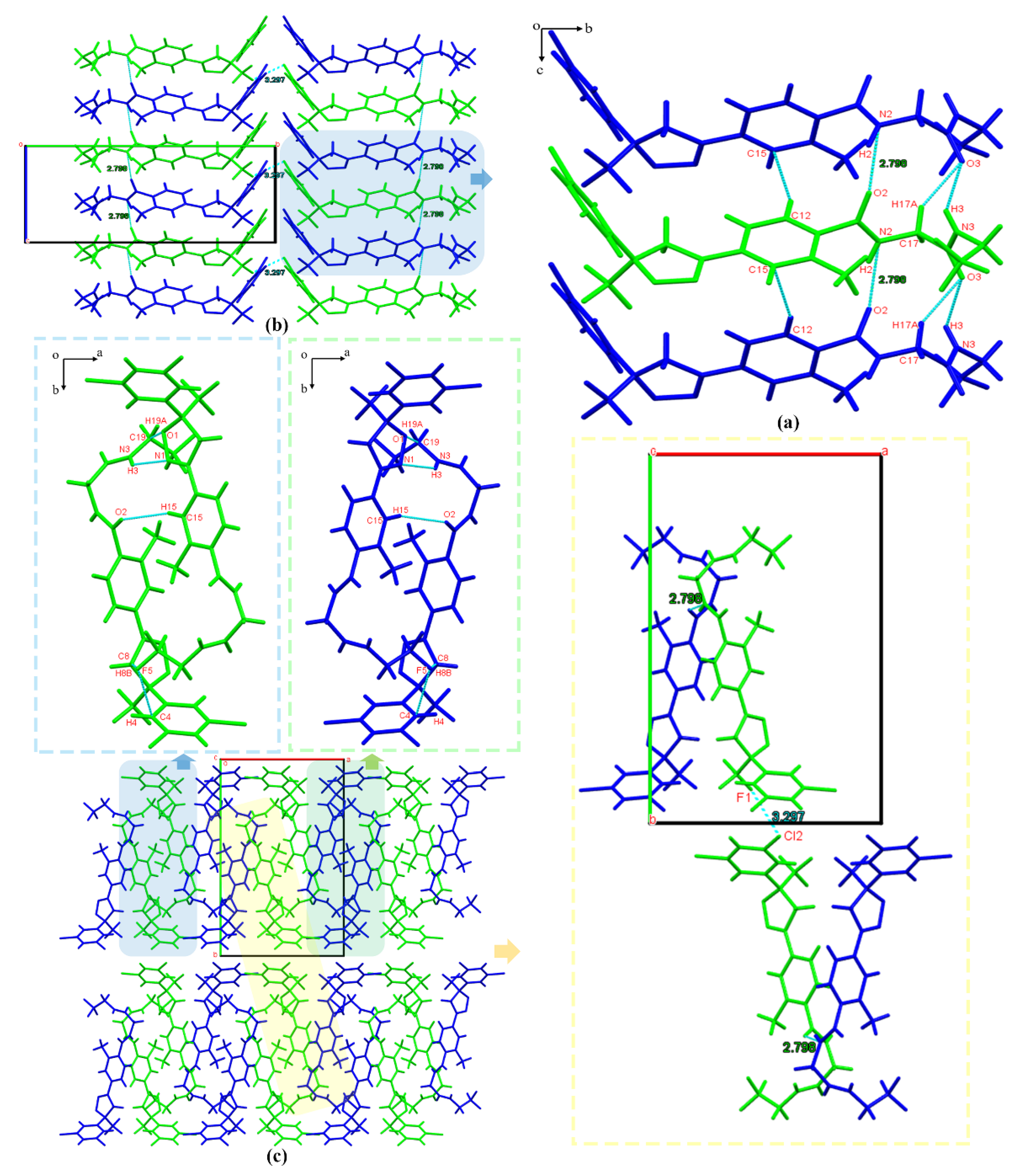
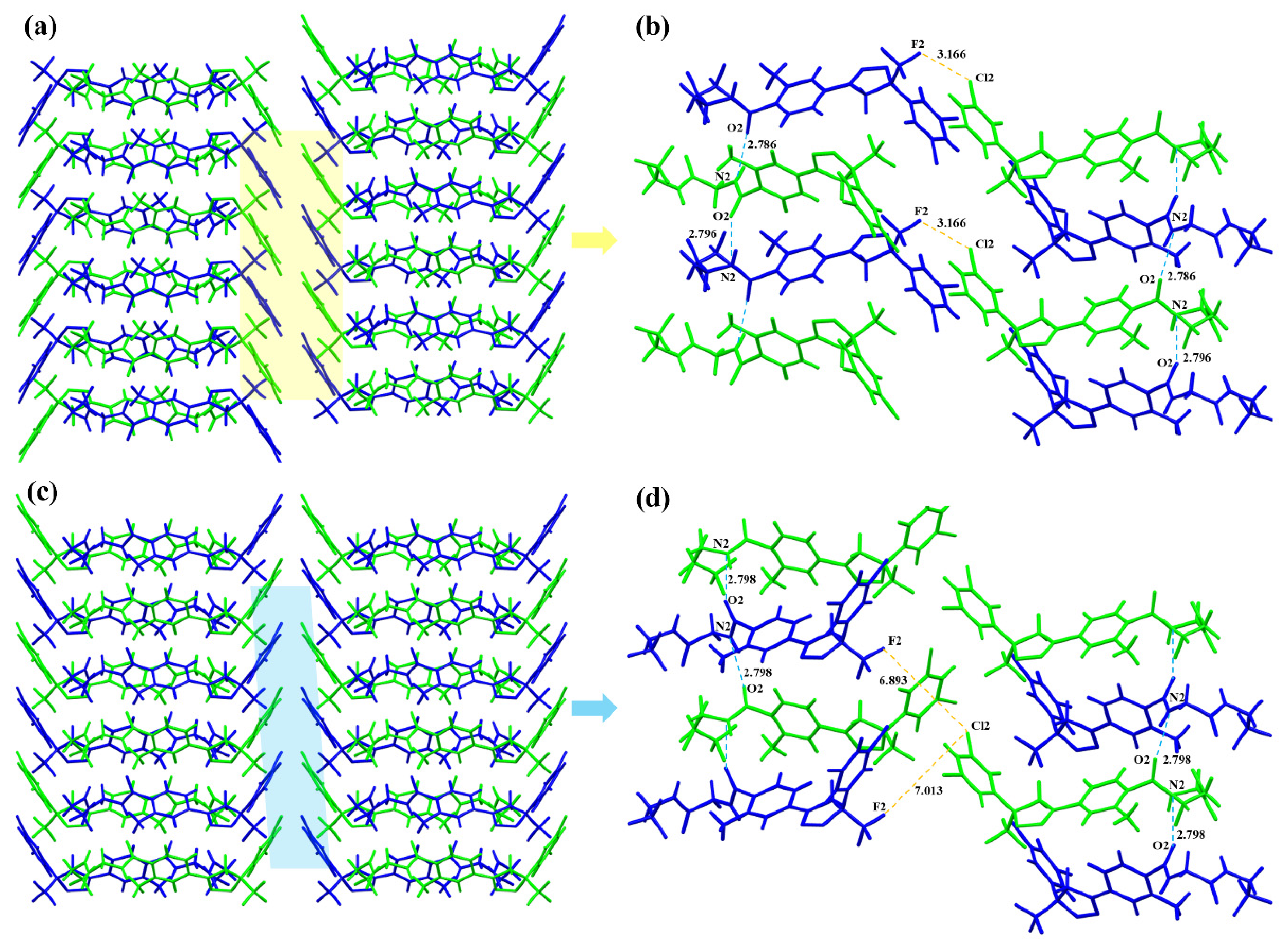
3.2.2. Crystal Structure Comparison
3.2.3. Molecular Conformations
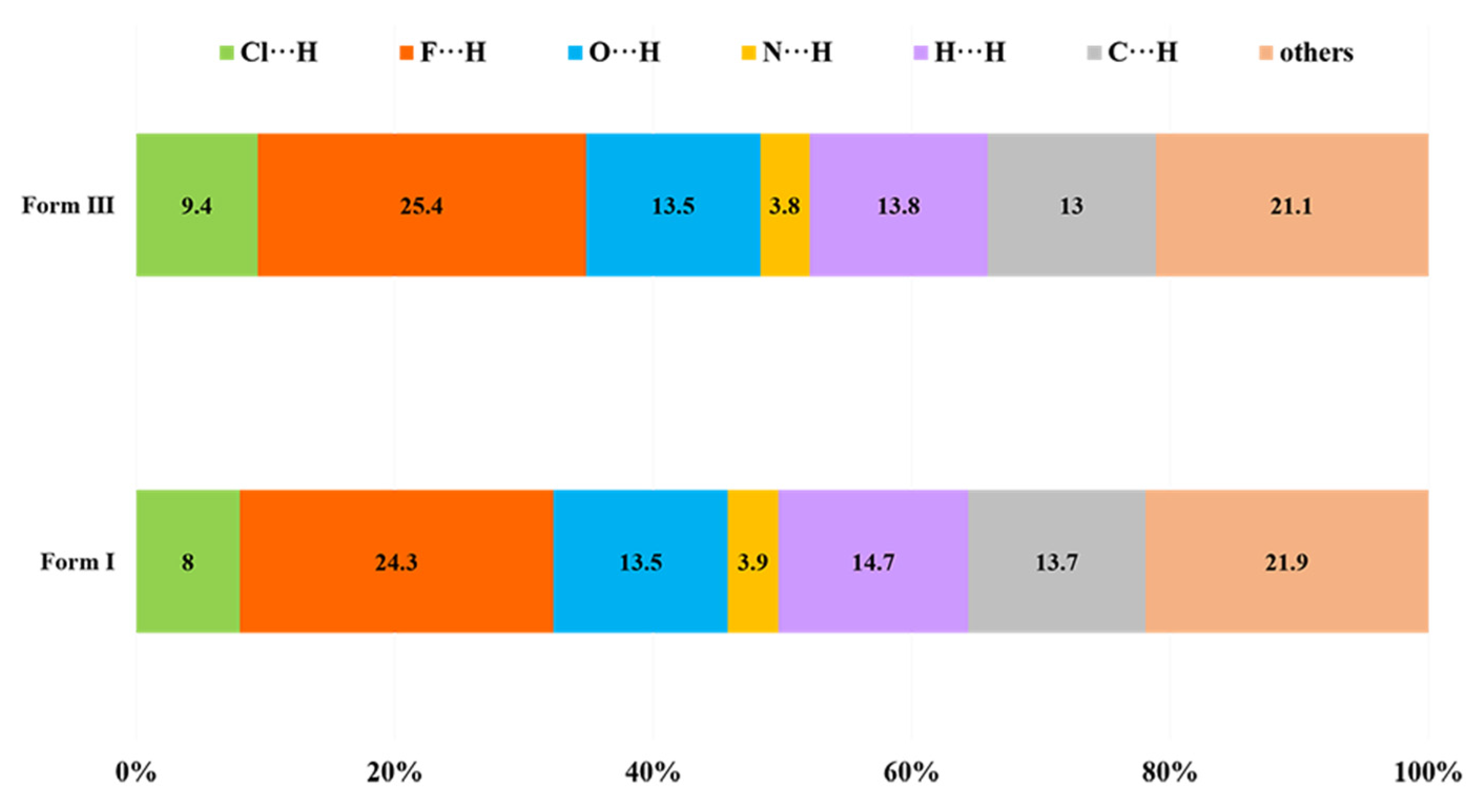
3.2.4. Hirshfeld Surface Analysis
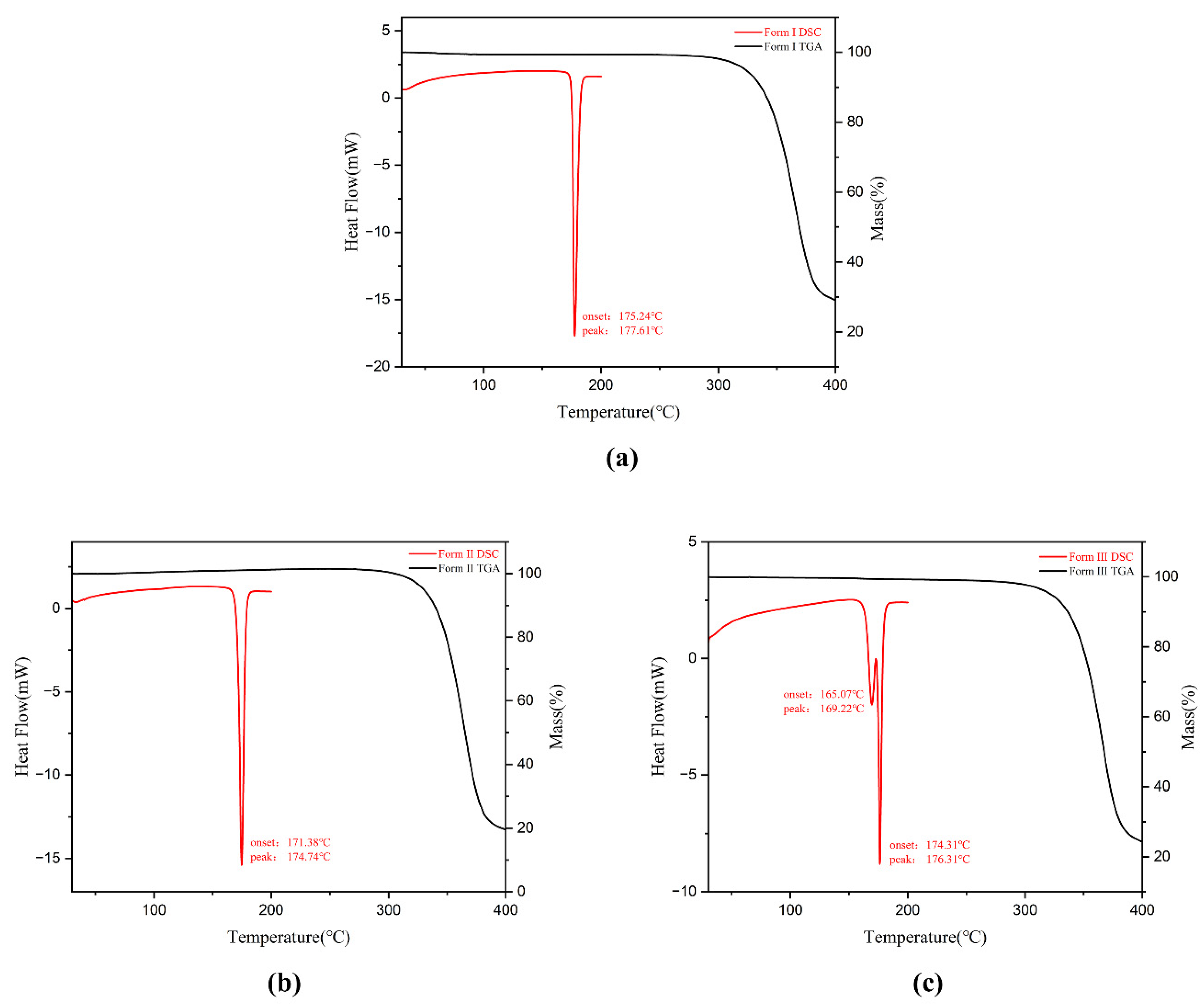
3.3. Thermal Analysis
3.4. Solvent-Mediated Phase Transformation
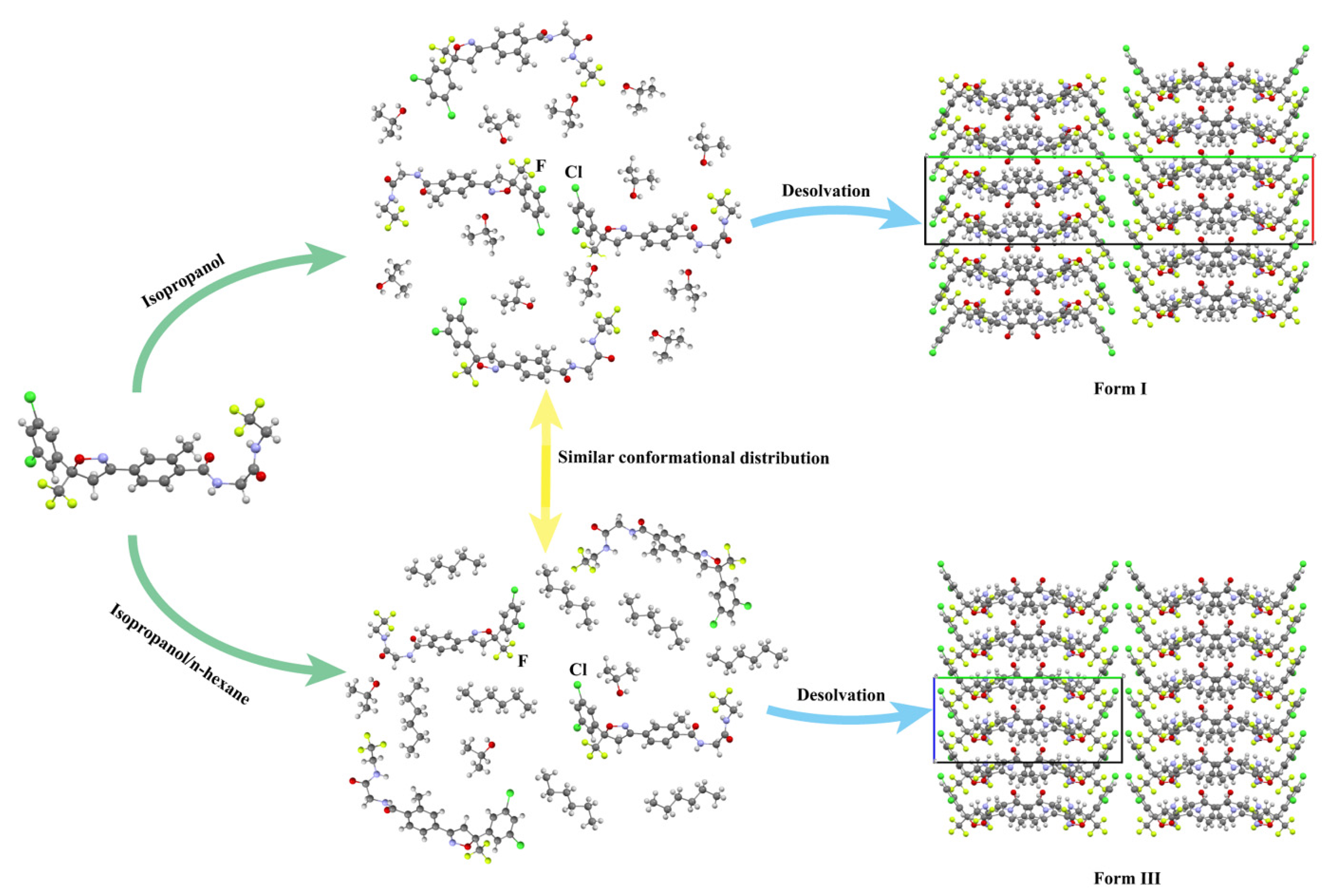
3.5. Selective Nucleation Studies
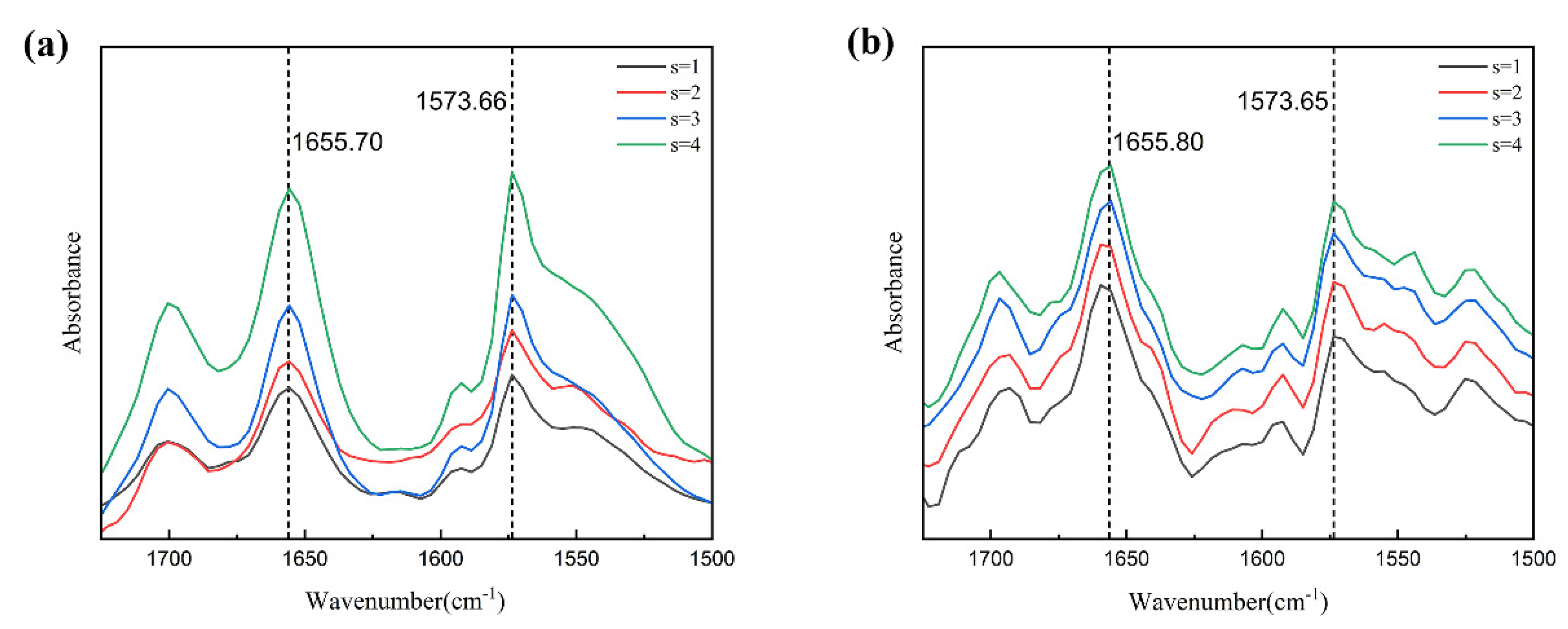
3.5.1. Solute–Solvent Interactions in Solution
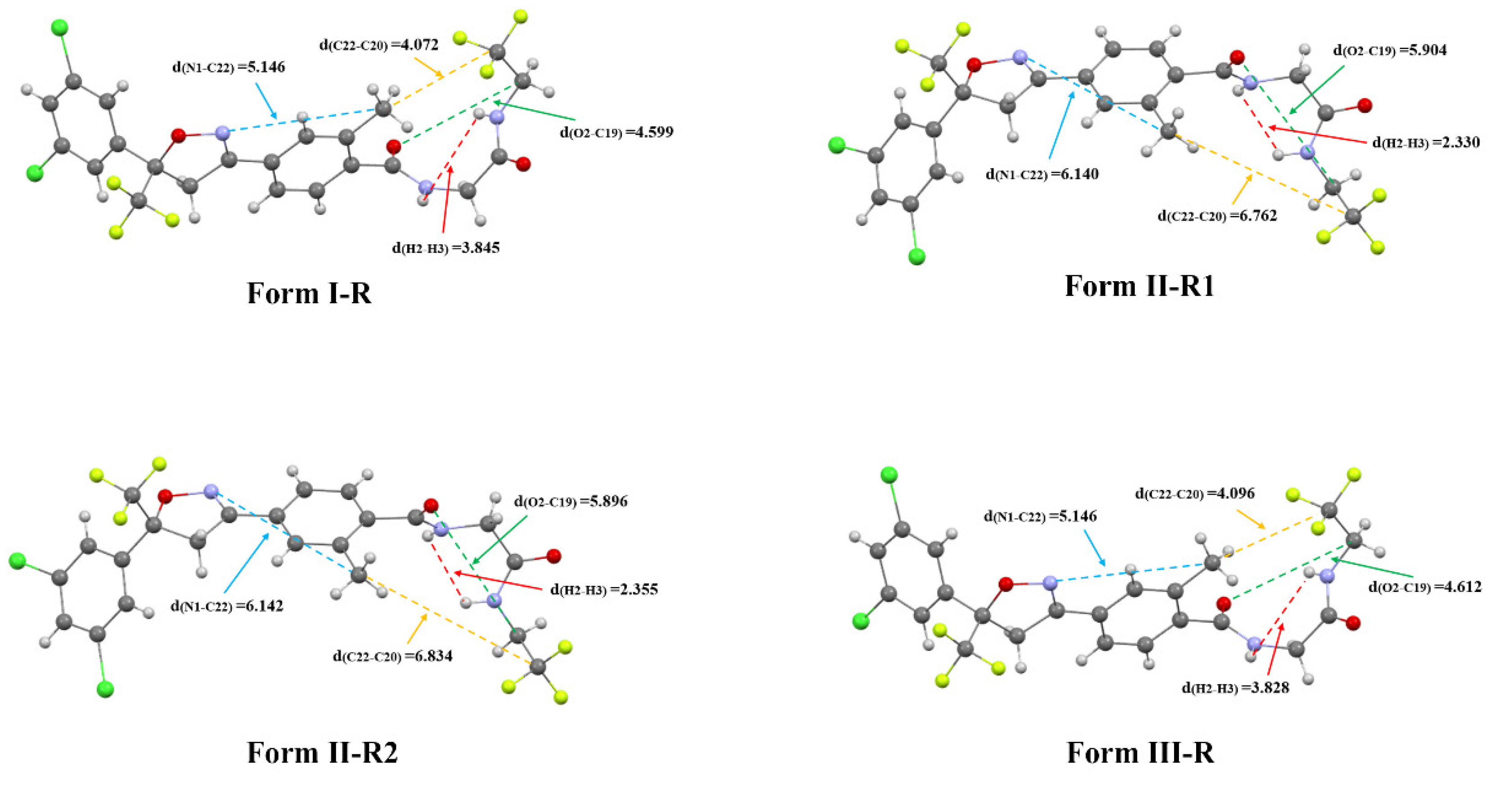
3.5.2. Conformational Distribution of Fluralaner Molecules in Solution
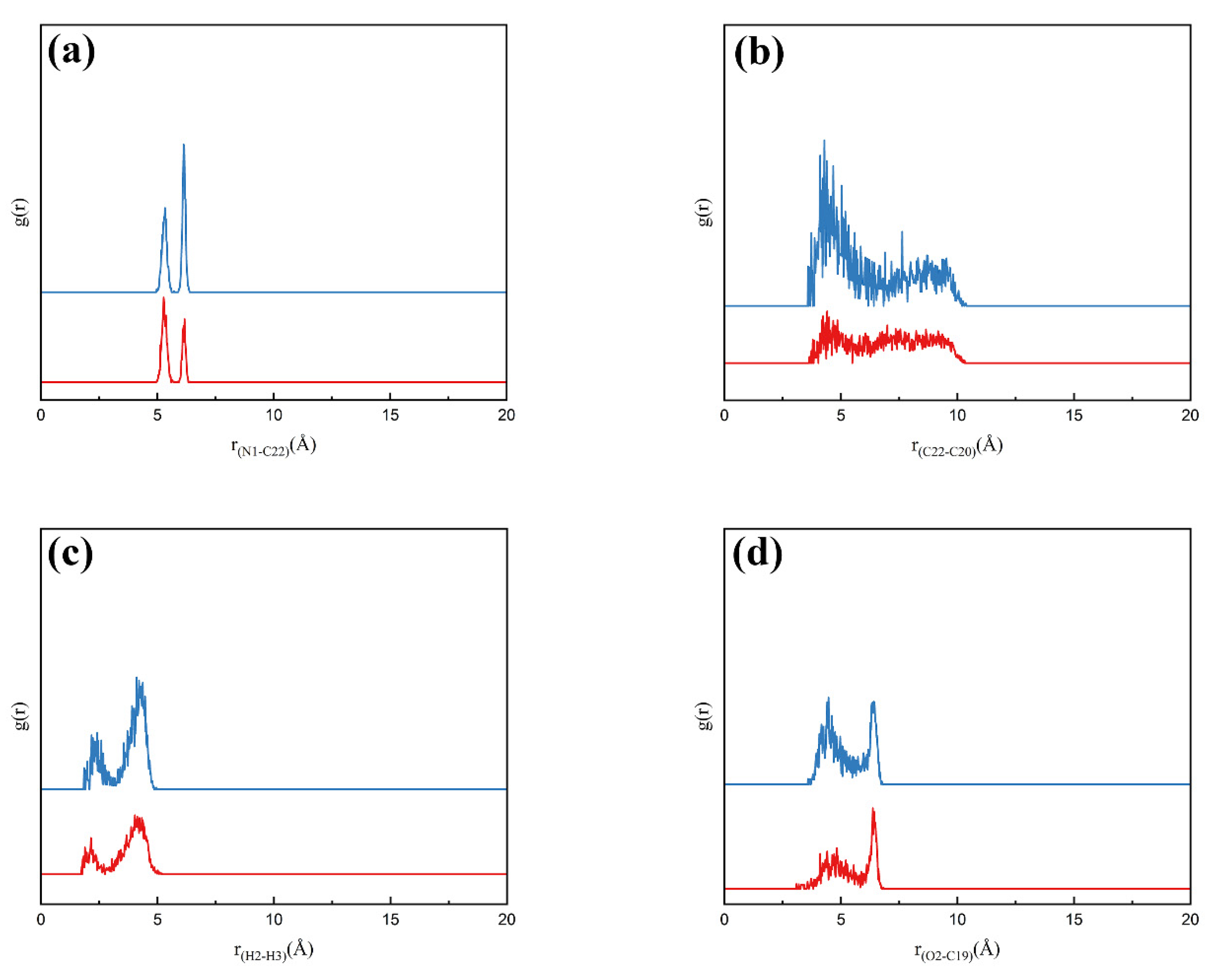
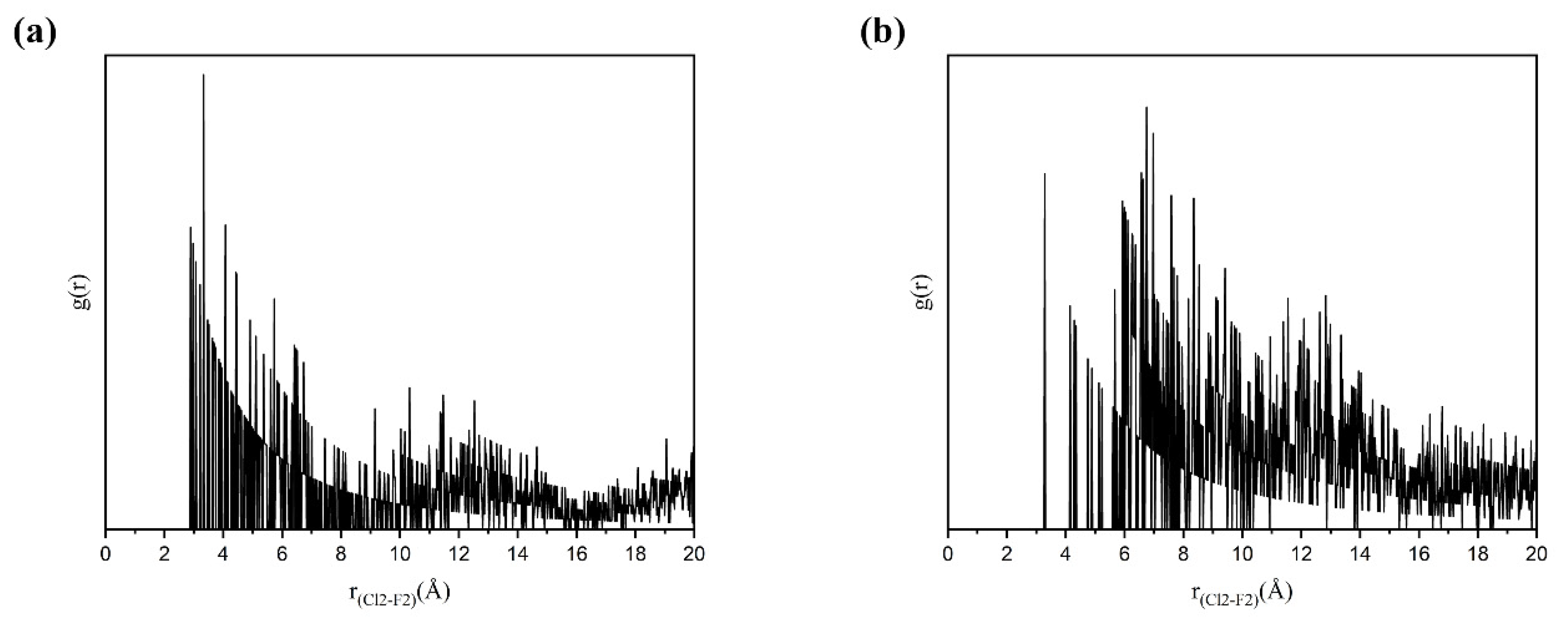
3.5.3. Solute–Solute Interactions in Solution
3.5.4. Selective Nucleation Mechanisms
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, B.; Wang, J.-R.; Mei, X. Insight into the Phase Transformation among Various Solid Forms of Baicalein. Cryst. Growth Des. 2015, 15, 4959–4968. [Google Scholar] [CrossRef]
- Wang, J.-R.; Zhu, B.; Yu, Q.; Mei, X. Selective crystallization of vitamin D3for the preparation of novel conformational polymorphs with distinctive chemical stability. CrystEngComm 2016, 18, 1101–1104. [Google Scholar] [CrossRef]
- Bommaka, M.K.; Chaitanya Mannava, M.K.; Rai, S.K.; Suresh, K.; Nangia, A.K. Entacapone Polymorphs: Crystal Structures, Dissolution, Permeability, and Stability. Cryst. Growth Des. 2021, 21, 5573–5585. [Google Scholar] [CrossRef]
- Fandaruff, C.; Rauber, G.S.; Araya-Sibaja, A.M.; Pereira, R.N.; de Campos, C.E.M.; Rocha, H.V.A.; Monti, G.A.; Malaspina, T.; Silva, M.A.S.; Cuffini, S.L. Polymorphism of Anti-HIV Drug Efavirenz: Investigations on Thermodynamic and Dissolution Properties. Cryst. Growth Des. 2014, 14, 4968–4975. [Google Scholar] [CrossRef]
- Bauer, J.; Spanton, S.; Henry, R.; Quick, J.; Dziki, W.; Porter, W.; Morris, J. Ritonavir: An extraordinary example of conformational polymorphism. Pharm. Res. 2001, 18, 859–866. [Google Scholar] [CrossRef]
- Ding, Z.; Su, W.; Huang, X.; Tian, B.; Cheng, X.; Mao, Y.; Li, G.; Liu, H.; Hao, H. Understanding the Role of Water in Different Solid Forms of Avibactam Sodium and Its Affecting Mechanism. Cryst. Growth Des. 2020, 20, 1150–1161. [Google Scholar] [CrossRef]
- Hao, H.; Barrett, M.; Hu, Y.; Su, W.; Ferguson, S.; Wood, B.; Glennon, B. The Use of in Situ Tools To Monitor the Enantiotropic Transformation of p-Aminobenzoic Acid Polymorphs. Org. Process. Res. Dev. 2011, 16, 35–41. [Google Scholar] [CrossRef]
- Li, S.; Wang, T.; Huang, X.; Zhou, L.; Chen, M.; Liu, W.; Zhang, X.; Dong, Y.; Hao, H. The role of water in the formation of crystal structures: A case study of valnemulin hydrochloride. CrystEngComm 2021, 23, 47–55. [Google Scholar] [CrossRef]
- Davey, R.J.; Schroeder, S.L.; ter Horst, J.H. Nucleation of organic crystals--a molecular perspective. Angew. Chem. Int. Ed. Engl. 2013, 52, 2166–2179. [Google Scholar] [CrossRef]
- Van Driessche, A.E.S.; Van Gerven, N.; Bomans, P.H.H.; Joosten, R.R.M.; Friedrich, H.; Gil-Carton, D.; Sommerdijk, N.; Sleutel, M. Molecular nucleation mechanisms and control strategies for crystal polymorph selection. Nature 2018, 556, 89–94. [Google Scholar] [CrossRef]
- Chen, J.; Trout, B.L. Computational study of solvent effects on the molecular self-assembly of tetrolic acid in solution and implications for the polymorph formed from crystallization. J. Phys. Chem. B 2008, 112, 7794–7802. [Google Scholar] [CrossRef] [PubMed]
- Davey, R.J.; Blagden, N.; Righini, S.; Alison, H.; Quayle, M.J.; Fuller, S. Crystal polymorphism as a probe for molecular self-assembly during nucleation from solutions: The case of 2,6-dihydroxybenzoic acid. Cryst. Growth Des. 2001, 1, 59–65. [Google Scholar] [CrossRef]
- Gaines, E.; Di Tommaso, D. Solvation and Aggregation of Meta-Aminobenzoic Acid in Water: Density Functional Theory and Molecular Dynamics Study. Pharmaceutics 2018, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, N.; Yang, J.; Huang, Y.; Ji, X.; Huang, X.; Wang, T.; Wang, H.; Hao, H. Molecular conformational evolution mechanism during nucleation of crystals in solution. IUCrJ 2020, 7, 542–556. [Google Scholar] [CrossRef]
- Zong, S.; Wang, J.K.; Wu, H.; Liu, Q.; Hao, Y.H.; Huang, X.; Wu, D.H.; Zhou, G.C.; Hao, H.X. Insight into the role of pre-assembly and desolvation in crystal nucleation: A case of p-nitrobenzoic acid. Acta Crystallogr. B 2019, 75, 845–854. [Google Scholar] [CrossRef]
- Du, W.; Cruz-Cabeza, A.J.; Woutersen, S.; Davey, R.J.; Yin, Q. Can the study of self-assembly in solution lead to a good model for the nucleation pathway? The case of tolfenamic acid. Chem. Sci. 2015, 6, 3515–3524. [Google Scholar] [CrossRef]
- Joseph, A.; Rodrigues Alves, J.S.; Bernardes, C.E.S.; Piedade, M.F.M.; Minas da Piedade, M.E. Tautomer selection through solvate formation: The case of 5-hydroxynicotinic acid. CrystEngComm 2019, 21, 2220–2233. [Google Scholar] [CrossRef]
- Burton, R.C.; Ferrari, E.S.; Davey, R.J.; Finney, J.L.; Bowron, D.T. The Relationship between Solution Structure and Crystal Nucleation: A Neutron Scattering Study of Supersaturated Methanolic Solutions of Benzoic Acid. J. Phys. Chem. B 2010, 114, 8807–8816. [Google Scholar] [CrossRef]
- Hylton, R.K.; Tizzard, G.J.; Threlfall, T.L.; Ellis, A.L.; Coles, S.J.; Seaton, C.C.; Schulze, E.; Lorenz, H.; Seidel-Morgenstern, A.; Stein, M.; et al. Are the Crystal Structures of Enantiopure and Racemic Mandelic Acids Determined by Kinetics or Thermodynamics? J. Am. Chem. Soc. 2015, 137, 11095–11104. [Google Scholar] [CrossRef]
- Mattei, A.; Li, T.L. Polymorph Formation and Nucleation Mechanism of Tolfenamic Acid in Solution: An Investigation of Pre-nucleation Solute Association. Pharm. Res. 2012, 29, 460–470. [Google Scholar] [CrossRef]
- de la Fuente, J.; Villar, M.; Contreras, M.; Moreno-Cid, J.A.; Merino, O.; Perez de la Lastra, J.M.; de la Fuente, G.; Galindo, R.C. Prospects for vaccination against the ticks of pets and the potential impact on pathogen transmission. Vet. Parasitol. 2015, 208, 26–29. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lehmann, T. Ectoparasites: Direct impact on host fitness. Parasitol. Today 1993, 9, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Sarmah, A.K.; Meyer, M.T.; Boxall, A.B. A global perspective on the use, sales, exposure pathways, occurrence, fate and effects of veterinary antibiotics (VAs) in the environment. Chemosphere 2006, 65, 725–759. [Google Scholar] [CrossRef] [PubMed]
- Hiroyuki, K.; Shunsuke, F.; Yuji, M.; Manabu, Y.; Takashi, M. Manufacturing Method of Isoxazoline Substituted Benzamide Compounds. WO Patent WO2010005048A1, 9 July 2011. [Google Scholar]
- Bongiorno, G.; Meyer, L.; Evans, A.; Lekouch, N.; Doherty, P.; Chiummo, R.; Gradoni, L. Insecticidal efficacy against Phlebotomus perniciosus in dogs treated orally with fluralaner in two different parallel-group, negative-control, random and masked trials. Parasites Vectors 2022, 15, 18. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, K.C.; Chiummo, R.M.; Heckeroth, A.R.; Zschiesche, E.; Brandao Guedes, P.E.; Harvey, T.V.; de Jesus, A.V.; da Paixao Seva, A.; de Oliveira, J.T.S.; Dos Santos Freire, Z.; et al. Efficacy of oral fluralaner (Bravecto) against Tunga penetrans in dogs: A negative control, randomized field study in an endemic community in Brazil. PLoS Negl. Trop. Dis. 2022, 16, e0010251. [Google Scholar] [CrossRef]
- Sari, A.B.; Gunes, Y.; Anlas, C.; Ustun Alkan, F.; Guncum, E.; Ustuner, O.; Bakirel, T. Effects of feed intake and water hardness on fluralaner pharmacokinetics in layer chickens. J. Vet. Sci. 2022, 23, e64. [Google Scholar] [CrossRef]
- Xiong, T.; Ling, S.Q.; Liu, J.L.; Zeng, X.N. Insecticidal and P450 mediate metabolism of fluralaner against red imported fire ant, Solenopsis invicta (Hymenoptera: Formicidae). Pestic. Biochem. Physiol. 2022, 187, 105184. [Google Scholar] [CrossRef]
- Yan, Y.; Feng, L.; Shi, M.; Cui, C.; Liu, Y. Effect of plasma-activated water on the structure and in vitro digestibility of waxy and normal maize starches during heat-moisture treatment. Food Chem. 2020, 306, 125589. [Google Scholar] [CrossRef]
- Ma, H.; Chen, S.; Song, Y.; Yin, D.; Li, X.; Li, X. Experimental investigation into the effects of composition and microstructure on the tensile properties and failure characteristics of different gypsum rocks. Sci. Rep. 2021, 11, 14517. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van De Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Martin, A.D.; Hartlieb, K.J.; Sobolev, A.N.; Raston, C.L. Hirshfeld Surface Analysis of Substituted Phenols. Cryst. Growth Des. 2010, 10, 5302–5306. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Fabbiani, F.P.A.; Spackman, M.A. Comparison of polymorphic molecular crystal structures through Hirshfeld surface analysis. Cryst. Growth Des. 2007, 7, 755–769. [Google Scholar] [CrossRef]
- Wang, Y.; Verma, P.; Jin, X.S.; Truhlar, D.G.; He, X. Revised M06 density functional for main-group and transition-metal chemistry. Proc. Natl. Acad. Sci. USA 2018, 115, 10257–10262. [Google Scholar] [CrossRef]
- Sun, H.; Jin, Z.; Yang, C.W.; Akkermans, R.L.C.; Robertson, S.H.; Spenley, N.A.; Miller, S.; Todd, S.M. COMPASS II: Extended coverage for polymer and drug-like molecule databases. J. Mol. Model. 2016, 22, 47. [Google Scholar] [CrossRef]
- Al-Zoubi, N.; Koundourellis, J.E.; Malamataris, S. FT-IR and Raman spectroscopic methods for identification and quantitation of orthorhombic and monoclinic paracetamol in powder mixes. J. Pharm. Anal. 2002, 29, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Palotai, R.; Nussinov, R. Induced fit, conformational selection and independent dynamic segments: An extended view of binding events. Trends Biochem. Sci. 2010, 35, 539–546. [Google Scholar] [CrossRef]
- Taylor, R.; Macrae, C.F. Rules governing the crystal packing of mono- and dialcohols. Acta Crystallogr. B 2001, 57, 815–827. [Google Scholar] [CrossRef]
- Tan, S.L.; Jotani, M.M.; Tiekink, E.R.T. Utilizing Hirshfeld surface calculations, non-covalent interaction (NCI) plots and the calculation of interaction energies in the analysis of molecular packing. Acta Crystallogr. Sect. E 2019, 75, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Jemai, M.; Khalfi, M.; Issaoui, N.; Roisnel, T.; Kazachenko, A.S.; Al-Dossary, O.; Marouani, H.; Malyar, Y.N. Role of Non-Covalent Interactions in Novel Supramolecular Compound, Bis(4-phenylpiperazin-1-ium) Oxalate Dihydrate: Synthesis, Molecular Structure, Thermal Characterization, Spectroscopic Properties and Quantum Chemical Study. Crystals 2023, 13, 875. [Google Scholar] [CrossRef]
- Das, D.; Banerjee, R.; Mondal, R.; Howard, J.A.; Boese, R.; Desiraju, G.R. Synthon evolution and unit cell evolution during crystallisation. A study of symmetry-independent molecules (Z′ > 1) in crystals of some hydroxy compounds. Chem. Commun. 2006, 5, 555–557. [Google Scholar] [CrossRef]
- Joseph, A.; Bernardes, C.E.S.; Druzhinina, A.I.; Varushchenko, R.M.; Nguyen, T.Y.; Emmerling, F.; Yuan, L.; Dupray, V.; Coquerel, G.; da Piedade, M.E.M. Polymorphic Phase Transition in 4′-Hydroxyacetophenone: Equilibrium Temperature, Kinetic Barrier, and the Relative Stability of Z′ = 1 and Z′ = 2 Forms. Cryst. Growth Des. 2017, 17, 1918–1932. [Google Scholar] [CrossRef]
- Desiraju, G.R. On the presence of multiple molecules in the crystal asymmetric unit (Z' > 1). CrystEngComm 2007, 9, 91–92. [Google Scholar] [CrossRef]
- Steed, K.M.; Steed, J.W. Packing problems: High Z' crystal structures and their relationship to cocrystals, inclusion compounds, and polymorphism. Chem. Rev. 2015, 115, 2895–2933. [Google Scholar] [CrossRef] [PubMed]
- Derdour, L.; Pack, S.K.; Skliar, D.; Lai, C.J.; Kiang, S. Crystallization from solutions containing multiple conformers: A new modeling approach for solubility and supersaturation. Chem. Eng. Sci. 2011, 66, 88–102. [Google Scholar] [CrossRef]
- Derdour, L.; Skliar, D. A review of the effect of multiple conformers on crystallization from solution and strategies for crystallizing slow inter-converting conformers. Chem. Eng. Sci. 2014, 106, 275–292. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymorph | Form I | Form II | Form III |
|---|---|---|---|
| Empirical formula | C22H17Cl2F6N3O3 | C22H17Cl2F6N3O3 | C22H17Cl2F6N3O3 |
| Formula weight | 556.28 | 556.28 | 556.28 |
| Temperature (K) | 113(2) | 295(9) | 113(2) |
| Crystal system | Monoclinic | Triclinic | Orthorhombic |
| Space group | P21/c | P | Pca21 |
| a (Å) | 9.0555(3) | 9.5129(3) | 12.6058(7) |
| b (Å) | 39.9230(9) | 15.1300(3) | 20.0622(15) |
| c (Å) | 12.6015(4) | 17.6150(5) | 9.0538(5) |
| α (deg) | 90 | 72.469(2) | 90 |
| β (deg) | 90.376(3) | 83.305(2) | 90 |
| γ (deg) | 90 | 89.077(2) | 90 |
| Cell volume (Å3) | 4555.6(2) | 2400.61(12) | 2289.7(2) |
| ρcalc (g/cm3) | 1.622 | 1.539 | 1.614 |
| Rint | 0.0550 | 0.0960 | 0.0943 |
| R1 (I > 2σ(I)) | 0.0563 | 0.0946 | 0.0518 |
| wR2 | 0.1481 | 0.2623 | 0.0866 |
| GOF(S) | 1.058 | 1.208 | 1.041 |
| CCDC | 2,253,035 | 2,253,036 | 2,253,037 |
| Form I | Form II (R1) | Form II (R2) | Form III | |
|---|---|---|---|---|
| Relative energy (kcal/mol) | 0 | 1.57 | 1.57 | 0.05 |
| Lattice Energy (kcal/mol) | 73.55 | 74.95 | 70.80 | |
| ΔGid | ΔGvdw | ΔGelec | ΔGsolv | |
|---|---|---|---|---|
| Isopropanol | −67.376 | −22.897 | 62.288 | −27.986 |
| Isopropanol/n-hexane | −67.258 | −21.437 | 65.642 | −23.053 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, G.; Wang, T.; Huang, X.; Wang, N.; Zhou, L.; Tian, B.; Feng, Z.; Liu, A.; Li, Y.; Hao, H. Crystal Structure Analysis, Stability, Phase Transformation and Selective Nucleation Mechanism of Fluralaner Polymorphs. Crystals 2023, 13, 1241. https://doi.org/10.3390/cryst13081241
Li G, Wang T, Huang X, Wang N, Zhou L, Tian B, Feng Z, Liu A, Li Y, Hao H. Crystal Structure Analysis, Stability, Phase Transformation and Selective Nucleation Mechanism of Fluralaner Polymorphs. Crystals. 2023; 13(8):1241. https://doi.org/10.3390/cryst13081241
Chicago/Turabian StyleLi, Guangyan, Ting Wang, Xin Huang, Na Wang, Lina Zhou, Beiqian Tian, Ziwei Feng, Ailing Liu, Yaling Li, and Hongxun Hao. 2023. "Crystal Structure Analysis, Stability, Phase Transformation and Selective Nucleation Mechanism of Fluralaner Polymorphs" Crystals 13, no. 8: 1241. https://doi.org/10.3390/cryst13081241
APA StyleLi, G., Wang, T., Huang, X., Wang, N., Zhou, L., Tian, B., Feng, Z., Liu, A., Li, Y., & Hao, H. (2023). Crystal Structure Analysis, Stability, Phase Transformation and Selective Nucleation Mechanism of Fluralaner Polymorphs. Crystals, 13(8), 1241. https://doi.org/10.3390/cryst13081241