
Structural and Electronic Properties of U5M+ and T5M+ (U = Uracil, T = Thymine, M = Ag and Au) Cluster Cations

Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
4. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Conflicts of Interest
References
- Davis, J.T. G-quartets 40 years later: From 5′-GMP to molecular biology and supramolecular chemistry. Angew. Chem. Int. Ed. 2004, 43, 668–698. [Google Scholar] [CrossRef] [PubMed]
- Gellert, M.; Lipsett, M.N.; Davies, D.R. Helix formation by guanylic acid. Proc. Natl. Acad. Sci. USA 1962, 48, 2013–2018. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.; Gilbert, W. A sodium-potassium switch in the formation of four-stranded G4-DNA. Nature 1990, 344, 410–414. [Google Scholar] [CrossRef] [PubMed]
- González-Rodríguez, D.; van Dongen, J.L.; Lutz, M.; Spek, A.L.; Schenning, A.P.; Meijer, E. G-quadruplex self-assembly regulated by Coulombic interactions. Nat. Chem. 2009, 1, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Sigel, A.; Sigel, H.; Sigel, R.K. Interplay between Metal Ions and Nucleic Acids. Metal Ions in Life Sciences; Springer: Berlin/Heidelberg, Germany, 2012; Volume 10, pp. 119–134. [Google Scholar]
- Marlow, A.L.; Davis, J.T. Self-assembled ionophores as phase transfer catalysts. Tetrahedron Lett. 1999, 40, 3539–3542. [Google Scholar] [CrossRef]
- Chaput, J.C.; Switzer, C. A DNA pentaplex incorporating nucleobase quintets. Proc. Natl. Acad. Sci. USA 1999, 96, 10614–10619. [Google Scholar] [CrossRef]
- Gu, J.; Leszczynski, J. Isoguanine complexes: Quintet versus tetrad. J. Phys. Chem. B 2003, 107, 6609–6613. [Google Scholar] [CrossRef]
- Gu, J.; Wang, J.; Leszczynski, J. Iso-guanine quintet complexes coordinated by mono valent cations (Na+, K+, Rb+, and Cs+). J. Comput. Chem. 2007, 28, 1790–1795. [Google Scholar] [CrossRef]
- Meyer, M.; Sühnel, J. Self-association of isoguanine nucleobases and molecular recognition of alkaline ions: Tetrad vs pentad structures. J. Phys. Chem. A 2003, 107, 1025–1031. [Google Scholar] [CrossRef]
- Qiu, B.; Liu, J.; Qin, Z.; Wang, G.; Luo, H. Quintets of uracil and thymine: A novel structure of nucleobase self-assembly studied by electrospray ionization mass spectrometry. Chem. Commun. 2009, 2009, 2863–2865. [Google Scholar] [CrossRef]
- Ding, Y.; Wang, X.; Li, D.; Xu, W. Real-Space Evidence of Trimeric, Tetrameric, and Pentameric Uracil Clusters Induced by Alkali Metals. J. Phys. Chem. C 2020, 124, 5257–5262. [Google Scholar] [CrossRef]
- Rajabi, K.; Gillis, E.A.L.; Fridgen, T.D. Structures of Alkali Metal Ion-Adenine Complexes and Hydrated Complexes by IRMPD Spectroscopy and Electronic Structure Calculations. J. Phys. Chem. A 2010, 114, 3449–3456. [Google Scholar] [CrossRef] [PubMed]
- Russo, N.; Toscano, M.; Grand, A. Bond Energies and Attachments Sites of Sodium and Potassium Cations to DNA and RNA Nucleic Acid Bases in the Gas Phase. J. Am. Chem. Soc. 2001, 123, 10272–10279. [Google Scholar] [CrossRef] [PubMed]
- Krasnokutski, S.A.; Lee, J.S.; Yang, D.-S. High-resolution electron spectroscopy and structures of lithium-nucleobase (adenine, uracil, and thymine) complexes. J. Chem. Phys. 2010, 132, 044304–044308. [Google Scholar] [CrossRef] [PubMed]
- Valdespino-Saenz, J.; Martinez, A. Theoretical Study of Neutral, Anionic, and Cationic Uracil−Ag and Uracil−Au Systems: Nonconventional Hydrogen Bonds. J. Phys. Chem. A 2008, 112, 2408–2414. [Google Scholar] [CrossRef]
- Vazquez, M.-V.; Martinez, A. Theoretical Study of Cytosine−Al, Cytosine−Cu and Cytosine−Ag (Neutral, Anionic and Cationic). J. Phys. Chem. A 2008, 112, 1033–1039. [Google Scholar] [CrossRef]
- Martinez, A. Theoretical study of guanine—Cu and uracil—Cu (neutral, anionic, and cationic). Is it possible to carry out a photoelectron spectroscopy experiment? J. Chem. Phys. 2005, 123, 024311–024319. [Google Scholar] [CrossRef]
- Soto-Verdugo, V.; Metiu, H.; Gwinn, E. The properties of small Ag clusters bound to DNA bases. J. Chem. Phys. 2010, 132, 195102–195110. [Google Scholar] [CrossRef]
- Kryachko, E.S.; Remacle, F. Complexes of DNA Bases and Gold Clusters Au3 and Au4 Involving Nonconventional N−H⋯Au Hydrogen Bonding. Nano Lett. 2005, 5, 735–739. [Google Scholar] [CrossRef]
- Martinez, A. Do Anionic Gold Clusters Modify Conventional Hydrogen Bonds? The Interaction of Anionic Aun (n = 2 − 4) with the Adenine-Uracil Base Pair. J. Phys. Chem. A 2009, 113, 1134–1140. [Google Scholar] [CrossRef]
- Rincon, E.; Yanez, M.; Toro-Labbe, A.; Mo, O. Effect of Ni (II), Cu (II) and Zn (II) association on the keto-enol tautomerism of thymine in the gas phasewz. Phys. Chem. Chem. Phys. 2007, 9, 2531–2537. [Google Scholar] [CrossRef]
- Šponer, J.; Sabat, M.; Burda, J.V.; Leszczynski, J.; Hobza, P.; Lippert, B. Metal ions in non-complementary DNA base pairs: An ab initio study of Cu(I), Ag(I), and Au(I) complexes with the cytosine-adenine base pair. J. Biol. Inorg. Chem. 1999, 4, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Šponer, J.; Šponer, J.E.; Gorb, L.; Leszczynski, J.; Lippert, B. Metal-Stabilized Rare Tautomers and Mispairs of DNA Bases: N6-Metalated Adenine and N4-Metalated Cytosine, Theoretical and Experimental Views. J. Phys. Chem. A 1999, 103, 11406–11413. [Google Scholar] [CrossRef]
- Rodgers, M.T.; Armentrout, P.B. Influence of d Orbital Occupation on the Binding of Metal Ions to Adenine. J. Am. Chem. Soc. 2002, 124, 2678–2691. [Google Scholar] [CrossRef] [PubMed]
- Boychuk, B.T.A.; Wetmore, S.D. Assessment of Density Functional Theory Methods for the Structural Prediction of Transition and Post-Transition Metal–Nucleic Acid Complexes. J. Chem. Theory Comput. 2023, 19, 5273–5288. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.-J.; Xu, H.-G.; Zheng, W.-J.; Li, J. Theoretical and experimental studies of the interactions between Au2− and nucleobases. Phys. Chem. Chem. Phys. 2014, 16, 2928–2935. [Google Scholar] [CrossRef]
- Cao, G.-J.; Xu, H.-G.; Li, R.-Z.; Zheng, W.-J. Hydrogen bonds in the nucleobase-gold complexes: Photoelectron spectroscopy and density functional calculations. J. Chem. Phys. 2012, 136, 014305. [Google Scholar] [CrossRef]
- Cao, G.-J.; Xu, H.-G.; Xu, X.-L.; Wang, P.; Zheng, W.-J. Photodissociation and density functional calculations of A2M+ and G2M+ (A = adenine, G = guanine, M = Cu, Ag, and Au) cluster ions. Int. J. Mass Spectrom. 2016, 407, 118–125. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.P.G.A.; Mennucci, B.; Petersson, G.A.; et al. GAUSSIAN 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Figgen, D.; Rauhut, G.; Dolg, M.; Stoll, H. Energy-consistent pseudopotentials for group 11 and 12 atoms: Adjustment to multi-configuration Dirac–Hartree–Fock data. Chem. Phys. 2005, 311, 227–244. [Google Scholar] [CrossRef]
- Peterson, K.A.; Puzzarini, C. Systematically convergent basis sets for transition metals. II. Pseudopotential-based correlation consistent basis sets for the group 11 (Cu, Ag, Au) and 12 (Zn, Cd, Hg) elements. Theor. Chem. Acc. 2005, 114, 283–296. [Google Scholar] [CrossRef]
- Cao, G.-J.; Zheng, W.-J. Structures, Stabilities, and Physicochemical Properties of Nucleobase Tautomers. Acta Phys.-Chim. Sin. 2013, 29, 2135–2147. [Google Scholar]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Guerra, C.F.; Snijders, J.; Te Velde, G.; Baerends, E. Towards an order-N DFT method. Theor. Chem. Acc. 1998, 99, 391–403. [Google Scholar]
- Baerends, E.J.; Ziegler, T.; Autschbach, J.; Bashford, D.; Bérces, A.; Bickelhaupt, F.M.; Bo, C.; Boerrigter, P.M.; Cavallo, L.; Chong, D.P.; et al. ADF, SCM, Theoretical Chemistry; Vrije Universiteit: Amsterdam, The Netherlands, 2013; Available online: http://www.scm.com (accessed on 1 January 2024).
- Van Lenthe, E.; Baerends, E.J. Optimized Slater-type basis sets for the elements 1–118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef]
- Cao, G.-J. A dinuclear Cu(i)-mediated complex: Theoretical studies of the G2Cu24+ cluster ion. J. Chem. Phys. 2018, 149, 144308. [Google Scholar] [CrossRef]
- Nir, E.; Plützer, C.; Kleinermanns, K.; de Vries, M. Properties of isolated DNA bases, base pairs and nucleosides examined by laser spectroscopy. Eur. Phys. J. D 2002, 20, 317–329. [Google Scholar] [CrossRef]
- Hopffgarten, M.V.; Frenking, G. Energy decomposition analysis. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 43–62. [Google Scholar] [CrossRef]
- Mitoraj, M.P.; Michalak, A.; Ziegler, T. A Combined Charge and Energy Decomposition Scheme for Bond Analysis. J. Chem. Theory Comput. 2009, 5, 962–975. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.-J.; Schwarz, W.H.E.; Li, J. An 18-Electron System Containing a Superheavy Element: Theoretical Studies of Sg@Au12. Inorg. Chem. 2015, 54, 3695–3701. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I. Charge, bond order and valence in the AB initio SCF theory. Chem. Phys. Lett. 1983, 97, 270–274. [Google Scholar] [CrossRef]
- Cao, G.-J.; Hou, H.-L. Dinuclear Metal-Mediated Guanine–Uracil Base Pairs: Theoretical Studies of GUM22+(M = Cu, Ag, and Au) Ions. J. Clust. Sci. 2019, 30, 439–448. [Google Scholar] [CrossRef]
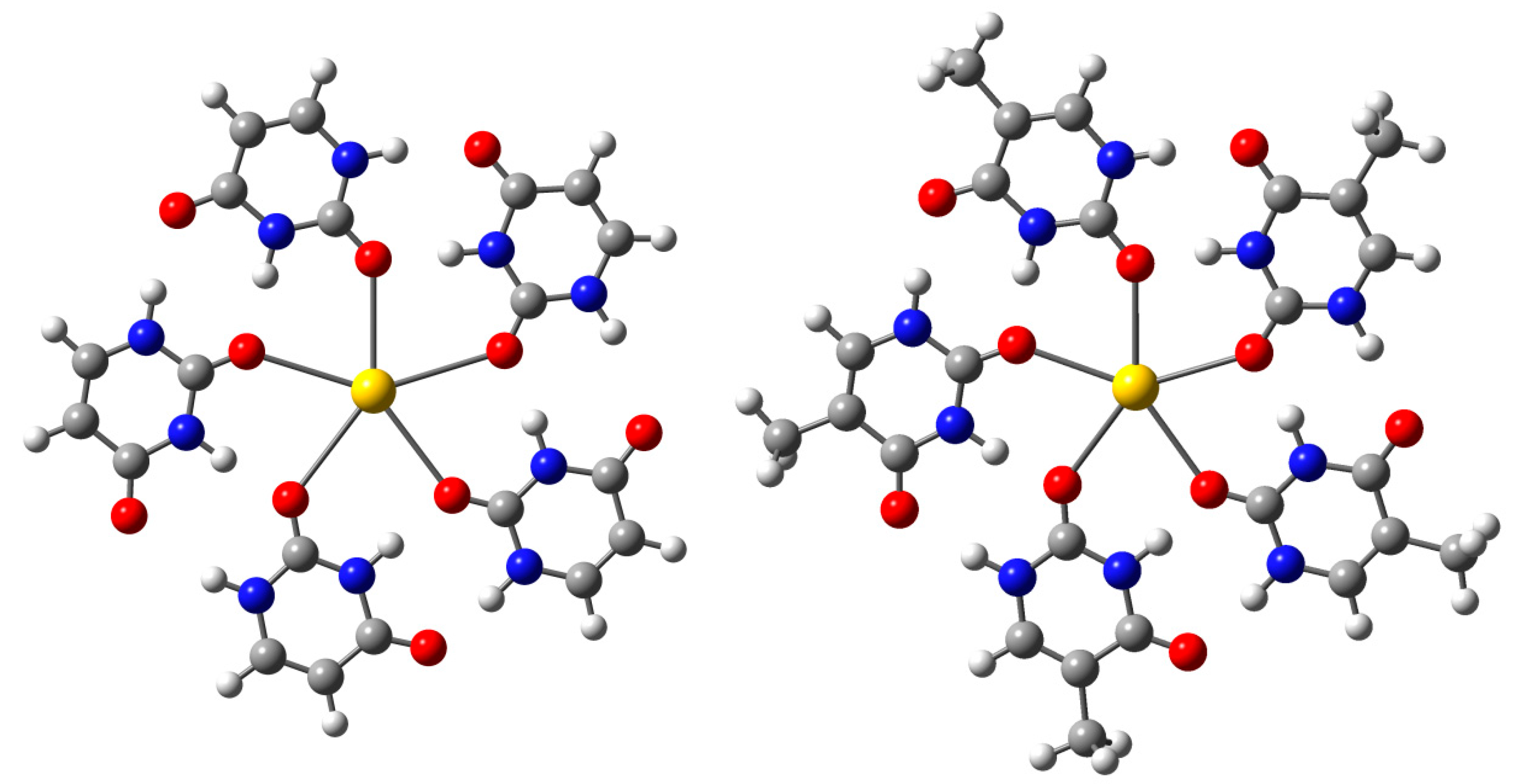
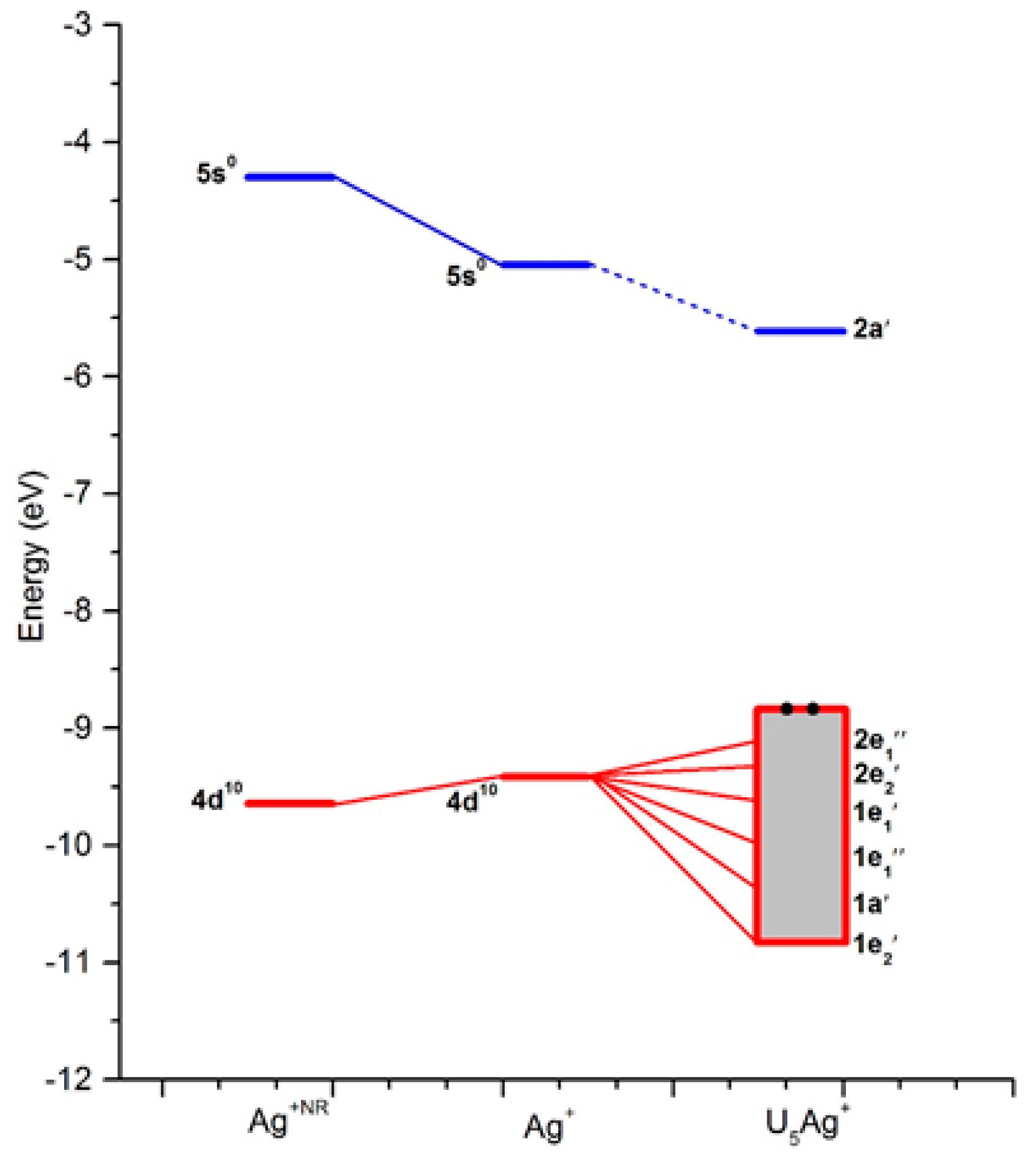
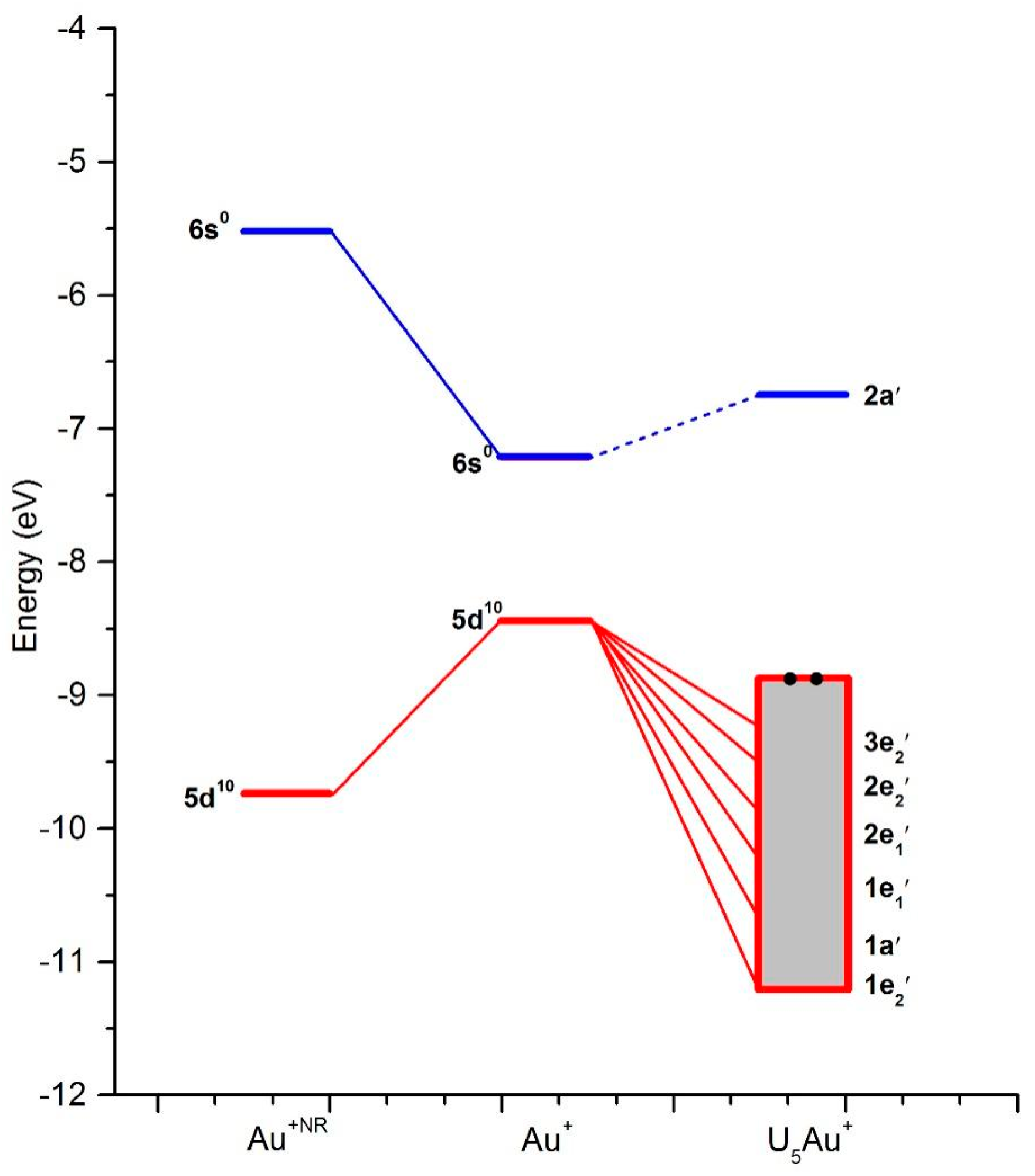

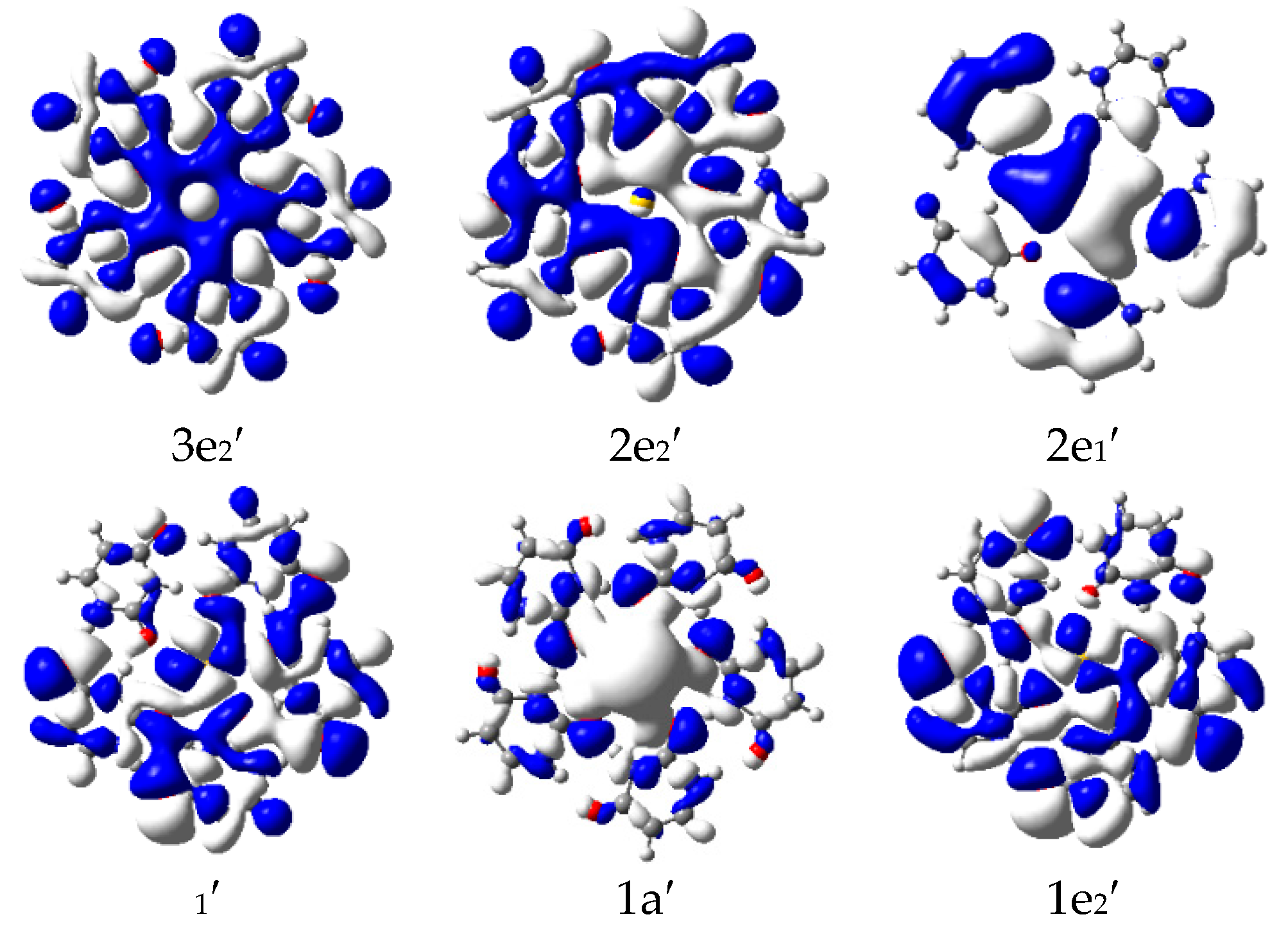

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M | Sym. | State | M-O Bond Lengths | Inner ∠N-H···O | Outer ∠N- H···O | |
|---|---|---|---|---|---|---|
| U5M+ | Ag | C5h | 1A′ | 2.737 | 172.3 | 173.3 |
| Au | C5h | 1A′ | 2.734 | 172.4 | 173.3 | |
| K | C5h | 1A′ | 2.859 | 173.7 | 172.2 | |
| Rb | C5h | 1A′ | 2.929 | 177.1 | 168.9 | |
| Cs | C5h | 1A′ | 3.036 | 179.8 | 166.3 | |
| T5M+ | Ag | C5h | 1A′ | 2.731 | 172.0 | 173.0 |
| Au | C5h | 1A′ | 2.727 | 172.0 | 173.1 | |
| K | C5h | 1A′ | 2.892 | 173.7 | 168.9 | |
| Rb | C5h | 1A′ | 2.923 | 176.5 | 168.6 | |
| Cs | C5h | 1A′ | 3.030 | 179.4 | 166.1 |
| M | Hirshfeld Charge | Voronoi Charge | |
|---|---|---|---|
| U5M+ | Ag | 0.49 | 0.43 |
| Au | 0.41 | 0.36 | |
| K | 0.48 | 0.28 | |
| Rb | 0.49 | 0.25 | |
| Cs | 0.50 | 0.19 | |
| T5M+ | Ag | 0.48 | 0.43 |
| Au | 0.40 | 0.36 | |
| K | 0.48 | 0.28 | |
| Rb | 0.49 | 0.25 | |
| Cs | 0.50 | 0.19 |
| MO | Type | ε | Occ | Ag(4d) | Ag(5s) |
|---|---|---|---|---|---|
| 1e2′ | Ag (d) + O (2p) | −10.8 | 4 | 41 | - |
| 1a′ | Ag (d) + O (2p) | −10.7 | 2 | 74 | - |
| 1e1″ | Ag (d) + O (2p) | −10.6 | 4 | 94 | - |
| 1e1′ | Ag (d) + O (2p) | −10.1 | 2 | 23 | 6 |
| 2e2′ | Ag (d) + O (2p) | −9.9 | 4 | 54 | - |
| 2e1″ | Ag (d) + O (2p) | −9.8 | 4 | 3 | - |
| 2a′ | Ag (Sd) + O (2p) | −5.6 | 0 | 1 | 91 |
| MO | Type | ε | Occ | Au(5d) | Au(6s) |
|---|---|---|---|---|---|
| 1e2″ | Au (d) + O (2p) | −10.8 | 4 | 19 | - |
| 1a′ | Au (d) + O (2p) | −10.5 | 2 | 12 | - |
| 1e1′ | Au (d) + O (2p) | −10.0 | 4 | 52 | - |
| 2e1′ | Au (d) + O (2p) | −9.5 | 4 | 44 | - |
| 2e2′ | Au (d) + O (2p) | −9.3 | 4 | 66 | - |
| 3e2′ | Au (d) + O (2p) | −8.9 | 4 | 10 | - |
| 2a′ | Au (Sd) + O (2p) | −6.8 | 0 | 2 | 82 |
| M | Steric Interaction | ∆EOrb | ∆EQuant | ∆EInt | |||
|---|---|---|---|---|---|---|---|
| ∆Eelstat | ∆EPauli | ∆ESter | |||||
| U5M+ | Ag | −2.15 (25.7%) | 3.23 | 1.08 | −6.20 (74.3%) | −2.97 | −5.12 |
| Au | −2.12 (31.5%) | 3.35 | 1.93 | −4.62 (68.5%) | −1.27 | −3.38 | |
| T5M+ | Ag | −2.19 (26.8%) | 3.28 | 1.09 | −5.99 (73.2%) | −2.71 | −4.89 |
| Au | −2.17 (32.9%) | 3.43 | 1.26 | −4.43 (67.1%) | −1.00 | −3.17 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, G.-J. Structural and Electronic Properties of U5M+ and T5M+ (U = Uracil, T = Thymine, M = Ag and Au) Cluster Cations. Crystals 2024, 14, 865. https://doi.org/10.3390/cryst14100865
Cao G-J. Structural and Electronic Properties of U5M+ and T5M+ (U = Uracil, T = Thymine, M = Ag and Au) Cluster Cations. Crystals. 2024; 14(10):865. https://doi.org/10.3390/cryst14100865
Chicago/Turabian StyleCao, Guo-Jin. 2024. "Structural and Electronic Properties of U5M+ and T5M+ (U = Uracil, T = Thymine, M = Ag and Au) Cluster Cations" Crystals 14, no. 10: 865. https://doi.org/10.3390/cryst14100865