

Relative Cooperative Effects of Non-Covalent Interactions on Hydrogen Bonds in Model Y…HCN/HNC…XF Trimers (Y = FB, OC, N2, CO, BF; XF = HF, LiF, BeF2, BF3, ClF, PH2F, SF2, SiH3F)
Abstract
:1. Introduction
- (i)
- to examine the relative changes in selected geometric and spectroscopic properties of model linear hydrogen-bonded Y…HCN/HNC dimers, where Y denotes the diatomic isomers BF, CO and N2. The strength of the Y…H hydrogen bond can be modulated by systematically varying the relative orientation of the Y dipole moment, from FB to OC to N2 (favorably aligned with the HCN/HNC dipole), to CO and BF (opposite direction to the HCN/HNC dipole),
- (ii)
- to compare the relative binding strengths and selected geometric and spectroscopic properties of model HCN…XF and HNC…XF dimers, where XF denotes a set of Lewis acids chosen such that each atom X represents a different group of the periodic table, and thus, a different type of noncovalent C…X or N…X intermolecular interaction. The intermolecular interactions can be broadly categorized as either (a) donor-acceptor, (b) H- or Li-bonded or (c) σ-hole bonded,
- (iii)
- to assess the cooperative effect of the Y…H and C/N…X noncovalent interactions in the Y…HCN…XF and Y…HNC…XF trimers, with HCN or HNC acting simultaneously as a Lewis acid (electron acceptor) and a Lewis base (electron donor).
2. Computational Methodology
3. Discussion
3.1. Y…HCN and Y…HNC Dimers
3.2. HCN…XF and HNC…XF Dimers
- (i)
- The H-N and H-F stretching modes in HNC…HF are coupled, leading to an anomalously small red shift of the H-N stretching mode and a large diminution of its infrared intensity (I/I0 < < 1). No other complex shows coupling of the intramolecular H-N/H-C and X-F vibrational modes or a decrease in their infrared intensities;
- (ii)
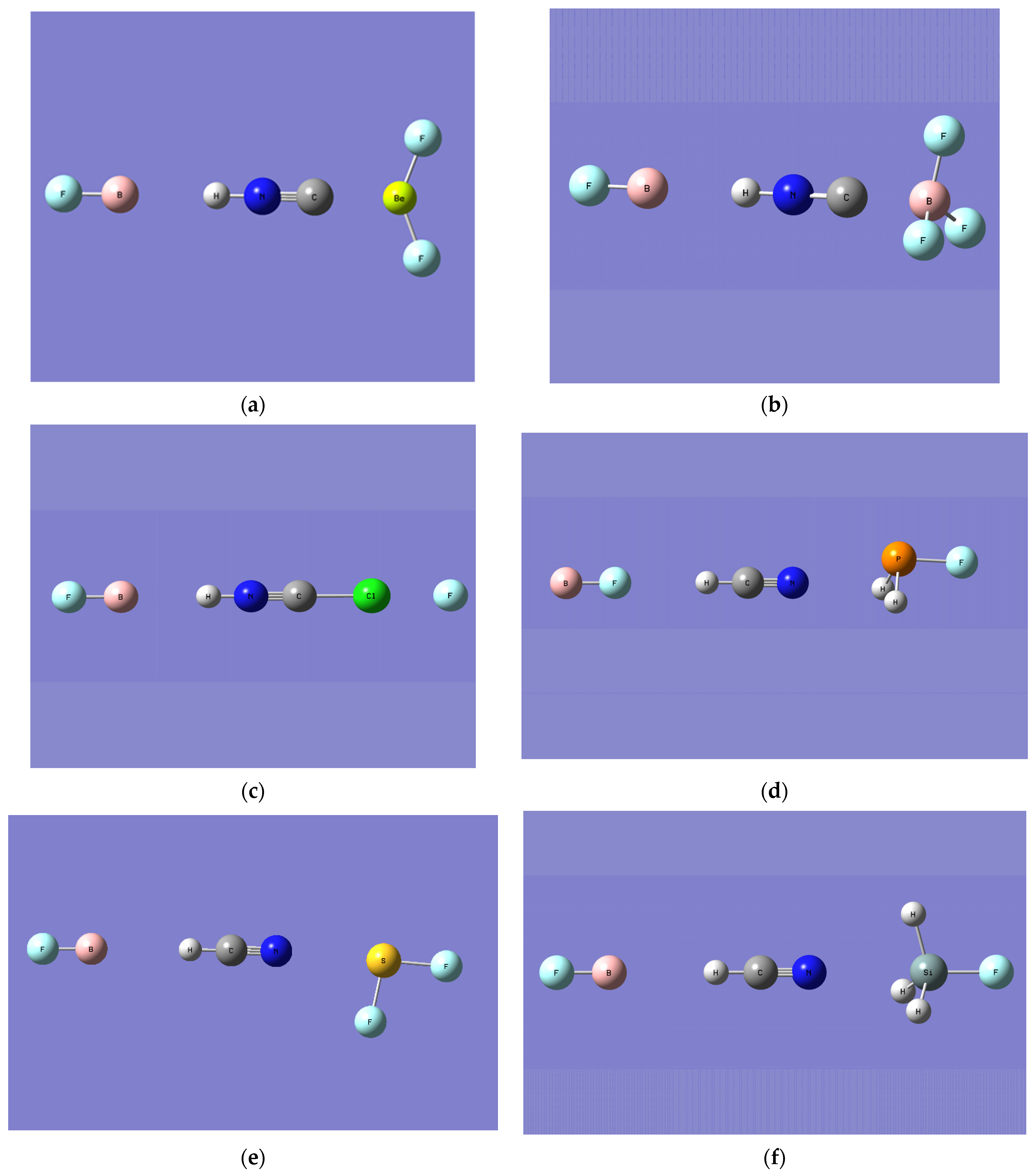
- HNC…BF3 is the second most strongly bound complex, with a C…B-F angle of 102°, indicating a 12°deviation from the planarity of the BF3 subunit on complexation with HNC (see Figure 1). By comparison, HCN…BF3 is more than 70 kJ/mol less strongly bound and the corresponding N…B-F angle is 93°, indicating a BF3 deviation of only 3° from planarity.
- (iii)
- HNC…ClF is much more strongly bound than HCN…ClF (by 61 kJ/mol) and the significant Cl-F bond extension of 0.2 Å suggests that the halogen bond between the monomers facilitates the transfer of the Cl from ClF to HNC; by contrast, there is little evidence of corresponding Cl transfer in HCN…ClF, where the Cl-F bond is elongated by only 0.018 Å.
3.3. YZ…HNC…XF Trimers
3.4. Y…HCN…XF Trimers
4. Conclusions
- (a)
- For the model Y…HCN/HNC dimers, the interaction energy increases systematically as the magnitude and orientation of the Y dipole, relative to the HCN/HNC molecular axis and dipole orientation were varied going from Y = BF to CO (opposite direction to the HCN or HNC dipole, thereby diminishing the intermolecular interaction), to the nonpolar N2, then to OC and FB (both in the same direction as the HCN or HNC dipole, thereby reinforcing the intermolecular interaction). The geometric and spectroscopic parameters also show a systematic trend with increased binding.We note the failure of the widely used M062x functional to correctly predict the H-C bond length change and frequency shift in the HCN complexes, which suggests that caution should be exercised in the use of this method for modeling complexes of HCN, and where feasible, validation by comparison with other methods, such as MP2, is advisable.
- (b)
- For the model HCN…XF and HNC…XF dimers, the interaction energy is generally in the order: donor-acceptor complexes (XF = BeF2, BF3) > H- or Li-bonded (HF, LiF) > σ-hole bonded complexes (ClF, PH2F, SF2, SiH3F). The energetic ordering for the σ-hole bonded complexes was found to be halogen > pnicogen > chalcogen > tetrel. The geometric and spectroscopic parameters for the H-C or H-N bonds in these complexes are well correlated with the binding strength, but the corresponding parameters for the X-F bond are less so. The infrared intensity enhancement ratio I/I0 for both uncomplexed and hydrogen-bonded H-N or H-C stretching modes in both dimers and trimers seems to be the most reliable molecular parameter for identifying trends with respect to the binding strength in both dimers and trimers containing HNC or HCN since it increases monotonically with increasing ΔE. For some of the systems studied here, this parameter may be relatively small, but nevertheless, usually shows a clear trend, especially for the σ-hole bonded complexes.
- (c)
- The FB/BF…HNC…XF trimers are more strongly bound than the FB/BF…HCN…XF trimers (as is the case for the corresponding HNC…XF and HCN…XF dimers) since HNC is more polar than HCN, but both series of complexes show similar trends for the corresponding geometric and spectroscopic parameters. Both sets of trimers also exhibit (positive) cooperative effects between the B/F…H hydrogen bond and the C/N…X noncovalent bond, with the nonadditive energies for FB…HCN/HNC…XF contributing between 7 and 19% to the total interaction energy, while the nonadditivity contribution to the total interaction energy in BF…HCN/HNC…XF is no greater than 5%, and so can be considered negligible.
- (d)
- The interaction energy and the nonadditive energy contribution to the trimer energetic stability correlate well with the relative strength of the noncovalent interactions due to XF, i.e., donor-acceptor > H- or Li-bonded > σ-hole bonded complexes. In other words, the B…H or F…H hydrogen bond is strengthened in the trimer by the XF interaction in accordance with the strength of the corresponding additional noncovalent bond. However, the N/C…X interaction is strengthened to a lesser extent by the B…H or F…H hydrogen bond, and has a more significant effect, on the relatively weaker XF noncovalent interactions.
- (e)
- As noted for the HCN/HNC…XF dimers, the infrared intensity enhancement of the H-N or H-C stretching mode appears to also be the molecular parameter that most closely correlates with the binding strength in the FB/BF…HCN/HNC…XF trimers, especially for the σ-hole bonded complexes. This parameter is probably most useful in distinguishing between closely related complexes, which may not be easily distinguishable by other spectroscopic approaches such as vibrational frequency shifts.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Müller-Dethlefs, K.; Hobza, P. Noncovalent interactions: A challenge for experiment and theory. Chem. Rev. 2000, 100, 143–168. [Google Scholar] [CrossRef] [PubMed]
- Hobza, P.; Zahradník, R.; Müller-Dethlefs, K. The world of non-covalent interactions: 2006. Collect. Czechoslov. Chem. Commun. 2006, 71, 443–531. [Google Scholar] [CrossRef]
- Schneider, H.-J. Noncovalent interactions: A brief account of a long history. J. Phys. Org. Chem. 2022, 35, e4340. [Google Scholar] [CrossRef]
- Scheiner, S. Hydrogen Bonding: A Theoretical Perspective; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Scheiner, S. Understanding noncovalent bonds and their controlling forces. J. Chem. Phys. 2020, 153, 140901. [Google Scholar] [CrossRef]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Gilli, G.; Gilli, P. Towards an unified hydrogen-bond theory. J. Mol. Struct. 2000, 552, 1–15. [Google Scholar] [CrossRef]
- Gilli, G.; Gilli, P. The Nature of the Hydrogen Bond: Outline of a Comprehensive Hydrogen Bond Theory; Oxford University Press: New York, NY, USA, 2009. [Google Scholar]
- Pimentel, G.C.; McClellan, A.L. The Hydrogen Bond; WH Freeman: San Francisco, CA, USA, 1960. [Google Scholar]
- Joseph, J.; Jemmis, E.D. Red-, blue-, or no-shift in hydrogen bonds: A unified explanation. J. Am. Chem. Soc. 2007, 129, 4620–4632. [Google Scholar] [CrossRef]
- Wang, C.; Danovich, D.; Shaik, S.; Mo, Y. A unified theory for the blue-and red-shifting phenomena in hydrogen and halogen bonds. J. Chem. Theory Comput. 2017, 13, 1626–1637. [Google Scholar] [CrossRef]
- Grabowski, S.J. What is the covalency of hydrogen bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef]
- Desiraju, G.R. Hydrogen bridges in crystal engineering: Interactions without borders. Acc. Chem. Res. 2002, 35, 565–573. [Google Scholar] [CrossRef]
- Jena, S.; Dutta, J.; Tulsiyan, K.D.; Sahu, A.K.; Choudhury, S.S.; Biswal, H.S. Noncovalent interactions in proteins and nucleic acids: Beyond hydrogen bonding and π-stacking. Chem. Soc. Rev. 2022, 51, 4261–4286. [Google Scholar] [CrossRef]
- Zhou, P.; Huang, J.; Tian, F. Specific noncovalent interactions at protein-ligand interface: Implications for rational drug design. Curr. Med. Chem. 2012, 19, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Lane, P.; Concha, M.C.; Ma, Y.; Murray, J.S. An overview of halogen bonding. J. Mol. Model. 2007, 13, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Brinck, T.; Murray, J.S.; Politzer, P. Surface electrostatic potentials of halogenated methanes as indicators of directional intermolecular interactions. Int. J. Quant. Chem. 1992, 44, 57–64. [Google Scholar] [CrossRef]
- Dong, W.; Li, Q.; Scheiner, S. Comparative Strengths of Tetrel, Pnicogen, Chalcogen, and Halogen Bonds and Contributing Factors. Molecules 2018, 23, 1681. [Google Scholar] [CrossRef] [PubMed]
- Aakeröy, C.B.; Fasulo, M.; Schultheiss, N.; Desper, J.; Moore, C. Structural competition between hydrogen bonds and halogen bonds. J. Am. Chem. Soc. 2007, 129, 13772–13773. [Google Scholar] [CrossRef] [PubMed]
- Solimannejad, M.; Malekani, M.; Alkorta, I. Cooperativity between the hydrogen bonding and halogen bonding in F3CX⋯NCH(CNH)⋯NCH(CNH) complexes (X=Cl, Br). Mol. Phys. 2011, 109, 1641–1648. [Google Scholar] [CrossRef]
- Scheiner, S. Sensitivity of noncovalent bonds to intermolecular separation: Hydrogen, halogen, chalcogen, and pnicogen bonds. CrystEngComm 2013, 15, 3119–3124. [Google Scholar] [CrossRef]
- Domagała, M.; Lutyńska, A.; Palusiak, M. Halogen bond versus hydrogen bond: The many-body interactions approach. Int. J. Quant. Chem. 2017, 117, e25348. [Google Scholar] [CrossRef]
- Robertson, C.C.; Wright, J.S.; Carrington, E.J.; Perutz, R.N.; Hunter, C.A.; Brammer, L. Hydrogen bonding vs. halogen bonding: The solvent decides. Chem. Sci. 2017, 8, 5392–5398. [Google Scholar] [CrossRef]
- Scheiner, S. The pnicogen bond: Its relation to hydrogen, halogen, and other noncovalent bonds. Acc. Chem. Res. 2013, 46, 280–288. [Google Scholar] [CrossRef]
- Scheiner, S. Detailed comparison of the pnicogen bond with chalcogen, halogen, and hydrogen bonds. Int. J. Quantum Chem. 2013, 113, 1609–1620. [Google Scholar] [CrossRef]
- Alkorta, I.; Del Bene, J.E.; Elguero, J. H2XP: OH2 Complexes: Hydrogen vs. pnicogen bonds. Crystals 2016, 6, 19. [Google Scholar] [CrossRef]
- Sethio, D.; Oliveira, V.; Kraka, E. Quantitative assessment of tetrel bonding utilizing vibrational spectroscopy. Molecules 2018, 23, 2763. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Li, Q. Interplay between tetrel bonding and hydrogen bonding interactions in complexes involving F2XO (X= C and Si) and HCN. Comput. Theor. Chem. 2014, 1050, 51–57. [Google Scholar] [CrossRef]
- Wang, X.; Li, Q.; Scheiner, S. Cooperativity between H-bonds and tetrel bonds. Transformation of a noncovalent C⋯N tetrel bond to a covalent bond. Phys. Chem. Chem. Phys. 2023, 25, 29738–29746. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Anderson, H.L. What is cooperativity? Angew. Chem. Int. Ed. Engl. 2009, 48, 7488. [Google Scholar] [CrossRef] [PubMed]
- Marin-Luna, M.; Alkorta, I.; Elguero, J. Cooperativity in tetrel bonds. J. Phys. Chem. A 2016, 120, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Sastry, G.N. Cooperative or anticooperative: How noncovalent interactions influence each other. J. Phys. Chem. B 2015, 119, 11121–11135. [Google Scholar] [CrossRef]
- Stone, A.J.; Buckingham, A.D.; Fowler, P.W. Comment on “Structure and spectroscopy of (HCN)n clusters: Cooperative and electronic delocalization effects in C–H⋯N hydrogen bonding” [J. Chem. Phys. 103, 333 (1995)]. J. Chem. Phys. 1997, 107, 1030–1031. [Google Scholar] [CrossRef]
- Li, Q.; Wang, X.; Cheng, J.; Li, W.; Gong, B.; Sun, J. Theoretical study on the interlay of hydrogen bonds in the trimers involving HCN and water. Int. J. Quantum Chem. 2009, 109, 1396–1402. [Google Scholar] [CrossRef]
- Kar, T.; Scheiner, S. Comparison of cooperativity in CH···O and OH···O hydrogen bonds. J. Phys. Chem. A 2004, 108, 9161–9168. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Behzadi, H.; Hadipour, N.L. Theoretical study of N–H··· O hydrogen bonding properties and cooperativity effects in linear acetamide clusters. Theor. Chem. Acc. 2008, 121, 135–146. [Google Scholar] [CrossRef]
- Henkel, S.; Misuraca, M.C.; Ding, Y.; Guitet, M.; Hunter, C.A. Enhanced chelate cooperativity in polar solvents. J. Am. Chem. Soc. 2017, 139, 6675–6681. [Google Scholar] [CrossRef] [PubMed]
- Frank, H.S.; Wen, W.-Y. Discuss. Faraday Soc. 1957, 24, 133. [Google Scholar] [CrossRef]
- Zeng, Y.; Hao, J.; Zheng, S.; Meng, L. Cooperativity between S···π and Rg···π in the OCS···C6H6···Rg (Rg = He, Ne, Ar, and Kr) van der Waals Complexes. J. Phys. Chem. A 2011, 115, 11057–11066. [Google Scholar] [CrossRef] [PubMed]
- Solimannejad, M.; Ghafari, S.; Esrafili, M.D. Theoretical insight into cooperativity in lithium-bonded complexes: Linear clusters of LiCN and LiNC. Chem. Phys. Lett. 2013, 577, 6–10. [Google Scholar] [CrossRef]
- Krishnapriya, V.U.; Suresh, C.H. Unraveling pnicogen bonding cooperativity: Insights from molecular electrostatic potential analysis. J. Comput. Chem. 2023; Early View. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Vakili, M.; Solimannejad, M. Cooperative effects in pnicogen bonding:(PH2F)2–7 and (PH2Cl)2–7 clusters. Chem. Phys. Lett. 2014, 609, 37–41. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Solimannejad, M. Single electron pnicogen bonded complexes. J. Phys. Chem. A 2014, 118, 947–953. [Google Scholar] [CrossRef]
- Nauta, K.; Miller, R.E. Nonequilibrium self-assembly of long chains of polar molecules in superfluid helium. Science 1999, 283, 1895–1897. [Google Scholar] [CrossRef]
- Dulmage, W.J.; Lipscomb, W.N. The crystal structures of hydrogen cyanide, HCN. Acta Crystallogr. 1951, 4, 330. [Google Scholar] [CrossRef]
- Matthews, C.N. The HCN world: Establishing protein—Nucleic acid life via hydrogen cyanide polymers. In Origins: Genesis, Evolution and Diversity of Life, Cellular Origin and Life in Extreme Habitats and Astrobiology; Springer: Dordrecht, The Netherlands, 2005; Volume 6, pp. 121–135. [Google Scholar]
- Sanchez, M.; Provasi, P.F.; Aucar, G.A.; Alkorta, I.; Elguero, J. Theoretical study of HCN and HNC neutral and charged clusters. J. Phys. Chem. B 2005, 109, 18189. [Google Scholar] [CrossRef] [PubMed]
- Brandão, I.; Rivelino, R.; Fonseca, T.L.; Castro, M.A. An ab initio study of electric properties of linear (HCN)N and (HNC)N aggregates in gas phase. Chem. Phys. Lett. 2013, 580, 9–13. [Google Scholar] [CrossRef]
- Adrian-Scotto, M.; Vasilescu, D. Density functional theory study of (HCN)n clusters up to n = 10. J. Mol. Struct. THEOCHEM 2007, 803, 45. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Uranyl Extraction by N, N-Dialkylamide Ligands Studied by Static and Dynamic DFT Simulations. In Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- McDowell, S.A.C.; Buckingham, A.D. Cooperative and diminutive hydrogen bonding in Y⋯ HCN⋯ HCN and NCH⋯ Y⋯ HCN trimers (Y= BF, CO, N2). J. Chem. Phys. 2010, 132, 064303. [Google Scholar] [CrossRef]
- Novak, A. Hydrogen bonding in solids correlation of spectroscopic and crystallographic data. In Large Molecules, Structure and Bonding; Springer: Berlin/Heidelberg, Germany, 2007; Volume 18, p. 177. [Google Scholar]
- Iogansen, A.V. Direct proportionality of the hydrogen bonding energy and the intensification of the stretching ν (XH) vibration in infrared spectra. Spectrochim. Acta Part A 1999, 55, 1585. [Google Scholar] [CrossRef]

{kind=link}
| Complex | ∆E | R (Y…H) | ∆r(H-N) or ∆r(H-C) | ∆ω(H-N) or ∆ω(H-C) | I/I0(H-N) or I/I0(H-C) |
|---|---|---|---|---|---|
| FB…H-CN | −13.7 (−12.4) | 2.606 (2.597) | 0.005 (−0.005) | −78.9 (15.2) | 3.65 (3.59) |
| OC…H-CN | −9.4 (−6.4) | 2.506 (2.592) | 0.003 (−0.008) | −38.8 (70.6) | 2.77 (2.33) |
| N2…H-CN | −6.8 (−4.5) | 2.461 (2.554) | 0.002 (−0.009) | −16.2 (92.4) | 2.25 (1.93) |
| CO…H-CN | −4.8 (−4.6) | 2.430 (2.458) | 0.001 (−0.010) | −3.7 (100.4) | 1.99 (1.82) |
| BF…H-CN | −2.8 (−2.4) | 2.428 (2.488) | 0.0 (−0.010) | 3.0 (105.1) | 1.70 (1.57) |
| FB…H-NC | −23.2 (−19.8) | 2.304 (2.322) | 0.012 (0.012) | −238.6 (−256.7) | 3.98 (3.59) |
| OC…H-NC | −15.2 (−10.8) | 2.220 (2.273) | 0.007 (0.006) | −131.7 (−125.6) | 2.97 (2.57) |
| N2…H-NC | −10.7 (−7.0) | 2.185 (2.258) | 0.003 (0.003) | −69.1 (−63.2) | 2.38 (2.12) |
| CO…H-NC | −7.8 (−6.5) | 2.173 (2.198) | 0.001 (0.002) | −31.5 (−47.8) | 2.08 (1.95) |
| BF…H-NC | −4.3 (−3.1) | 2.214 (2.217) | 0.0 (0.001) | −11.0 (−24.8) | 1.73 (1.66) |
| Complex | ∆E | R (N/C…X) | <N/C…X-F | ∆r(X-F) | ∆ω(H-C/N) | I/I0 (H-C/N)) | ∆ω(X-F) | I/I0 (X-F) |
|---|---|---|---|---|---|---|---|---|
| H-CN…BeF2 | −102.3 | 1.824 | 109 | 0.034 | −5.0 | 1.56 | ||
| H-CN…LiF | −67.8 | 2.073 | 180 | 0.021 | −10.7 | 1.39 | −5.5 | 1.35 |
| H-CN…HF | −33.2 | 1.853 | 180 | 0.012 | −2.0 | 1.36 | −269.3 | 6.91 |
| H-CN…BF3 | −30.3 | 2.460 | 93 | 0.004 | −2.0 | 1.24 | ||
| H-CN…ClF | −24.2 | 2.575 | 180 | 0.018 | −2.7 | 1.36 | −36.8 | 3.31 |
| H-CN…PH2F | −19.6 | 2.831 | 163 | 0.014 | −1.8 | 1.29 | −32.7 | 1.48 |
| H-CN…SF2 | −18.8 | 2.838 | 178 | 0.014 | −2.5 | 1.24 | −14.6 c | 1.20 |
| H-CN…SiH3F | −17.6 | 2.920 | 180 | 0.011 | −2.5 | 1.23 | −24.1 | 1.76 |
| H-NC…BeF2 | −112.9 | 1.914 | 110 | 0.037 | −29.8 | 1.46 | ||
| H-NC…BF3 | −104.3 | 1.843 | 102 | 0.043 | −27.1 | 1.55 | ||
| H-NC…ClF b | −85.1 | 1.760 | 180 | 0.217 | −29.8 | 2.50 | −255.9 | 11.4 |
| H-NC…LiF | −68.9 | 2.207 | 180 | 0.022 | −28.3 | 1.29 | −11.0 | 1.28 |
| H-NC…HF a | −36.2 | 1.925 | 180 | 0.016 | −7.8 | 0.06 | −368.7 | 10.3 |
| H-NC…PH2F | −22.7 | 2.762 | 166 | 0.019 | −12.2 | 1.31 | −49.1 | 1.74 |
| H-NC…SF2 | −20.2 | 2.862 | 178 | 0.018 | −11.3 | 1.25 | −22.4 c | 1.24 |
| H-NC…SiH3F | −18.4 | 2.962 | 180 | 0.013 | −10.7 | 1.22 | −29.9 | 1.94 |
| Complex | ∆E | Enonadd | R(Y…H) | ∆r(H-N) | ∆ω(H-N) | I/I0(H-N) | R(C…X) | ∆r(X-F) | ∆ω(X-F) | I/I0(X-F) |
|---|---|---|---|---|---|---|---|---|---|---|
| FB…HNC | −23.2 | 2.304 | 0.012 | −238.6 | 3.98 | |||||
| FB…HNC…BF3 | −158.0 | −30.5 | 2.129 | 0.027 | −489.9 | 7.55 | 1.791 | 0.050 | ||
| FB…HNC…BeF2 | −155.1 | −19.0 | 2.150 | 0.025 | −459.1 | 6.88 | 1.894 | 0.040 | ||
| FB…HNC…ClF a | −132.6 | −24.3 | 2.115 | 0.028 | −526.5 | 11.8 | 1.746 | 0.240 | −276.4 | 13.8 |
| FB…HNC…LiF | −104.0 | −11.9 | 2.182 | 0.022 | −411.0 | 5.97 | 2.189 | 0.026 | −15.4 | 1.35 |
| FB…HNC…HF | −65.4 | −6.0 | 2.234 | 0.017 | −326.7 | 6.01 | 1.896 | 0.019 | −423.6 | 8.00 |
| FB…HNC…PH2F | −52.9 | −7.0 | 2.249 | 0.016 | −303.7 | 5.61 | 2.706 | 0.024 | −63.1 | 1.99 |
| FB…HNC…SF2 | −47.9 | −4.5 | 2.251 | 0.015 | −300.4 | 5.41 | 2.784 | 0.024 | −26.3 | 1.39 |
| FB…HNC…SiH3F | −46.1 | −4.5 | 2.257 | 0.015 | −293.0 | 5.18 | 2.896 | 0.016 | −38.7 | 2.27 |
| BF…HNC | −4.3 | 2.214 | 0.0 | −11.0 | 1.73 | |||||
| BF…HNC…BeF2 | −120.0 | −2.8 | 2.084 | 0.004 | −59.7 | 2.70 | 1.910 | 0.037 | ||
| BF…HNC…BF3 | −113.4 | −4.8 | 2.074 | 0.004 | −59.1 | 2.89 | 1.834 | 0.044 | ||
| BF…HNC…ClF a | −92.7 | −3.3 | 2.072 | 0.004 | −64.5 | 4.46 | 1.758 | 0.220 | −258.8 | 12.5 |
| BF…HNC…LiF | −74.9 | −1.7 | 2.110 | 0.004 | −54.7 | 2.38 | 2.205 | 0.022 | −11.1 | 1.34 |
| BF…HNC…HF b | −41.2 | −0.7 | 2.158 | 0.002 | −18.9 | 0.27 | 1.921 | 0.016 | −382.2 | 12.3 |
| BF…HNC…PH2F | −27.7 | −0.7 | 2.171 | 0.002 | −26.4 | 2.30 | 2.754 | 0.019 | −50.3 | 1.82 |
| BF…HNC…SF2 | −25.2 | −0.7 | 2.173 | 0.001 | −25.3 | 2.21 | 2.849 | 0.018 | −23.0 | 1.26 |
| BF…HNC…SiH3F | −23.4 | −0.7 | 2.176 | 0.001 | −24.2 | 2.14 | 2.961 | 0.013 | −30.0 | 2.02 |
| Complex | ∆E | Enonadd | R(Y…H) | ∆r(H-C) | ∆ω(H-C) | I/I0(H-C) | R(N…X) | ∆r(X-F) | ∆ω(X-F) | I/I0(X-F) |
|---|---|---|---|---|---|---|---|---|---|---|
| FB…HCN | −13.7 | 2.606 | 0.005 | −78.9 | 3.65 | |||||
| FB…HCN…BeF2 | −129.2 | −13.2 | 2.444 | 0.011 | −156.7 | 6.89 | 1.805 | 0.036 | ||
| FB…HCN…LiF | −89.6 | −8.1 | 2.476 | 0.011 | −145.5 | 5.96 | 2.059 | 0.024 | −8.6 | 1.43 |
| FB…HCN…HF | −50.8 | −3.9 | 2.538 | 0.008 | −107.1 | 5.27 | 1.828 | 0.014 | −307.9 | 7.97 |
| FB…HCN…BF3 | −50.3 | −6.3 | 2.543 | 0.007 | −104.9 | 4.95 | 2.364 | 0.007 | ||
| FB…HCN…ClF | −41.2 | −3.3 | 2.549 | 0.007 | −103.7 | 5.31 | 2.531 | 0.022 | −44.1 | 4.01 |
| FB…HCN…PH2F | −35.8 | −2.5 | 2.565 | 0.007 | −97.9 | 4.91 | 2.794 | 0.016 | −37.8 | 1.59 |
| FB…HCN…SF2 | −34.9 | −2.4 | 2.566 | 0.007 | −97.1 | 4.78 | 2.801 | 0.016 | −16.6 | 1.18 |
| FB…HCN…SiH3F | −33.8 | −2.5 | 2.568 | 0.007 | −96.3 | 4.70 | 2.878 | 0.013 | −29.2 | 1.95 |
| BF…HCN | −2.9 | 2.428 | 0.0 | 3.0 | 1.70 | |||||
| BF…HCN…BeF2 | −106.6 | −1.4 | 2.296 | 0.002 | −7.4 | 2.84 | 1.822 | 0.034 | ||
| BF…HCN…LiF | −71.7 | −1.0 | 2.318 | 0.002 | −11.4 | 2.52 | 2.071 | 0.021 | −5.8 | 1.41 |
| BF…HCN…HF | −36.4 | −0.3 | 2.375 | 0.001 | −0.9 | 2.34 | 1.849 | 0.012 | −273.4 | 7.26 |
| BF…HCN…BF3 | −33.8 | −0.6 | 2.379 | 0.001 | 0.1 | 2.16 | 2.451 | 0.004 | ||
| BF…HCN…ClF | −27.4 | −0.3 | 2.384 | 0.001 | −1.4 | 2.33 | 2.567 | 0.019 | −37.3 | 3.51 |
| BF…HCN…PH2F | −22.7 | −0.2 | 2.392 | 0.001 | −1.2 | 2.21 | 2.822 | 0.014 | −32.1 | 1.52 |
| BF…HCN…SF2 | −21.9 | −0.2 | 2.382 | 0.001 | −1.4 | 2.17 | 2.824 | 0.015 | −13.3 | 1.11 |
| BF…HCN…SiH3F | −20.7 | −0.2 | 2.397 | 0.001 | −0.8 | 2.11 | 2.916 | 0.011 | −24.3 | 1.82 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McDowell, S.A.C.; Edwards, K.A. Relative Cooperative Effects of Non-Covalent Interactions on Hydrogen Bonds in Model Y…HCN/HNC…XF Trimers (Y = FB, OC, N2, CO, BF; XF = HF, LiF, BeF2, BF3, ClF, PH2F, SF2, SiH3F). Crystals 2024, 14, 111. https://doi.org/10.3390/cryst14020111
McDowell SAC, Edwards KA. Relative Cooperative Effects of Non-Covalent Interactions on Hydrogen Bonds in Model Y…HCN/HNC…XF Trimers (Y = FB, OC, N2, CO, BF; XF = HF, LiF, BeF2, BF3, ClF, PH2F, SF2, SiH3F). Crystals. 2024; 14(2):111. https://doi.org/10.3390/cryst14020111
Chicago/Turabian StyleMcDowell, Sean A. C., and Kodi A. Edwards. 2024. "Relative Cooperative Effects of Non-Covalent Interactions on Hydrogen Bonds in Model Y…HCN/HNC…XF Trimers (Y = FB, OC, N2, CO, BF; XF = HF, LiF, BeF2, BF3, ClF, PH2F, SF2, SiH3F)" Crystals 14, no. 2: 111. https://doi.org/10.3390/cryst14020111