
Ab Initio Study on the Vibrational and Electronic Properties of Radiation-Induced Defects in Potassium Bromide
 , , , and
, , , and
Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2. Computational Methods
3. Results and Discussion
3.1. Perfect KBr
3.2. Hole-Type Defects
3.3. Vacancy Defects
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kirkland, J. Quantitative application of potassium bromide disk technique in infrared spectroscopy. Anal. Chem. 1955, 27, 1537–1541. [Google Scholar] [CrossRef]
- Radhakrishna, S.; Chowdari, B.V. Radiation damage products in ionic crystals. Impurity doped alkali halides. Fortschr. Phys. 1977, 25, 511–578. [Google Scholar] [CrossRef]
- Ghomi, M.; Buisson, J. Study of Raman spectra of KBr doped with F-centres by excitation in the F and K bands. J. Phys. C Solid State Phys. 1979, 12, 4631. [Google Scholar] [CrossRef]
- Rzepka, E.; Doualan, J.L.; Lefrant, S.; Taurel, L. Raman scattering induced by V centres in KBr. J. Phys. C Solid State Phys. 1982, 15, L119–L123. [Google Scholar] [CrossRef]
- Suzuki, T.; Tanimura, K.; Itoh, N. Resonance-Raman-scattering spectroscopy for the halogen-molecular-ion centers in alkali halides. Phys. Rev. B 1993, 48, 9298–9305. [Google Scholar] [CrossRef] [PubMed]
- Taurel, L.; Rzepka, E.; Lefrant, S. Raman scattering induced by V centers and their aggregates in alkali halides. Radiat. Eff. 1983, 72, 115–121. [Google Scholar] [CrossRef]
- Popov, A.; Plavina, I. Photostimulated emission of KBr—In previously exposed to UV-or X-radiation. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 1995, 101, 252–254. [Google Scholar] [CrossRef]
- Rogulis, U.; Tale, I.; Hangleiter, T.; Spaeth, J.M. The photostimulation process in the X-ray storage phosphor KBr: In. J. Phys. Condens. Matter 1995, 7, 3129. [Google Scholar] [CrossRef]
- Okada, G.; Fujimoto, Y.; Tanaka, H.; Kasap, S.; Yanagida, T. Dynamics of radio-photoluminescence and thermally-stimulated luminescence in KBr: Sm. J. Mater. Sci. Mater. Electron. 2017, 28, 15980–15986. [Google Scholar] [CrossRef]
- Bridges, F.; Davies, G.; Robertson, J.; Stoneham, A. The spectroscopy of crystal defects: A compendium of defect nomenclature. J. Phys. Condens. Matter 1990, 2, 2875. [Google Scholar] [CrossRef]
- Itoh, N.; Tanimura, K. Formation of interstitial-vacancy pairs by electronic excitation in pure ionic crystals. J. Phys. Chem. Solids 1990, 51, 717–735. [Google Scholar] [CrossRef]
- Song, K.S.; Williams, R.T.; Halides, A. Self-Trapped Excitons; Springer: Berlin/Heidelberg, Germany, 1993; pp. 123–219. [Google Scholar]
- Saidoh, M.; Townsend, P. Absorption bands induced in KBr by H+ and H2+ implantation. J. Phys. C Solid State Phys. 1977, 10, 1541. [Google Scholar] [CrossRef]
- Akilbekov, A.; Elango, A. Low-Temperature Pair Associates of H Centres in KBr. Phys. Status Solidi (b) 1984, 122, 715–723. [Google Scholar] [CrossRef]
- Kuzovkov, V.; Popov, A.; Kotomin, E.; Moskina, A.; Vasilchenko, E.; Lushchik, A. Theoretical analysis of the kinetics of low-temperature defect recombination in alkali halide crystals. Low Temp. Phys. 2016, 42, 588–593. [Google Scholar] [CrossRef]
- Dauletbekova, A.; Akilbekov, A.; Elango, A. Thermo-and Photostimulated Recombinations of F–HA and α–IA Centres in KBr with Large Na Concentration. Phys. Status Solidi (b) 1982, 112, 445–452. [Google Scholar] [CrossRef]
- Akilbekov, A.; Dauletbekova, A.; Elango, A. Photo-and Thermochemical Reactions with Participation of Br- Centres in X-Rayed KBr. Phys. Status Solidi (b) 1985, 127, 493–501. [Google Scholar] [CrossRef]
- Popov, A. Optical production and destruction of V2 centres in KBr-In and KBr-T1 crystals. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 1992, 65, 521–524. [Google Scholar] [CrossRef]
- Lushchik, C.; Elango, A.; Gindina, R.; Pung, L.; Lushchik, A.; Maaroos, A.; Nurakhmetov, T.; Ploom, L.; Jaanson, N. Mechanisms of cation defects creation in alkali halides. Semicond. Insul. 1980, 5, 133–152. [Google Scholar]
- Nakajima, A.; Yoshihara, A.; Ishigame, M. Defect-induced Raman spectra in doped CeO2. Phys. Rev. B 1994, 50, 13297. [Google Scholar] [CrossRef]
- Ishigame, M.; Yoshida, E. Study of the defect-induced Raman spectra in cubic zirconia. Solid State Ionics 1987, 23, 211–218. [Google Scholar] [CrossRef]
- Bae, S.; Sugiyama, N.; Matsuo, T.; Raebiger, H.; Shudo, K.i.; Ohno, K. Defect-induced vibration modes of Ar+-irradiated MoS2. Phys. Rev. Appl. 2017, 7, 024001. [Google Scholar] [CrossRef]
- Akilbekov, A.; Kiryakov, A.; Baubekova, G.; Aralbayeva, G.; Dauletbekova, A.; Akylbekova, A.; Ospanova, Z.; Popov, A.I. Optical Characteristics of MgAl2O4 Single Crystals Irradiated by 220 MeV Xe Ions. Materials 2023, 16, 6414. [Google Scholar] [CrossRef]
- Wang, Y.; Pochet, P.; Jenkins, C.A.; Arenholz, E.; Bukalis, G.; Gemming, S.; Helm, M.; Zhou, S. Defect-induced magnetism in graphite through neutron irradiation. Phys. Rev. B 2014, 90, 214435. [Google Scholar] [CrossRef]
- Khomich, A.A.; Khmelnitsky, R.A.; Khomich, A.V. Probing the nanostructure of neutron-irradiated diamond using Raman spectroscopy. Nanomaterials 2020, 10, 1166. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Huang, L.; Zhu, W.; Ruan, Y. Raman scattering of neutron irradiated 6H-SiC. Solid State Commun. 2012, 152, 887–890. [Google Scholar] [CrossRef]
- Mironova-Ulmane, N.; Sildos, I.; Vasil’chenko, E.; Chikvaidze, G.; Skvortsova, V.; Kareiva, A.; Muñoz-Santiuste, J.; Pareja, R.; Elsts, E.; Popov, A. Optical absorption and Raman studies of neutron-irradiated Gd3Ga5O12 single crystals. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2018, 435, 306–312. [Google Scholar] [CrossRef]
- Platonenko, A.; Gryaznov, D.; Popov, A.I.; Dovesi, R.; Kotomin, E.A. First principles calculations of the vibrational properties of single and dimer F-type centers in corundum crystals. J. Chem. Phys. 2020, 153, 134107. [Google Scholar] [CrossRef]
- Rusevich, L.; Kotomin, E.; Zvejnieks, G.; Popov, A. Ab initio calculations of structural, electronic and vibrational properties of BaTiO3 and SrTiO3 perovskite crystals with oxygen vacancies. Low Temp. Phys. 2020, 46, 1185–1195. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R. Development of the Colle-Salvetti Correlation-Energy Formula Into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. Wires Comput. Mol. Sci. 2018, 8, e1360. Available online: https://onlinelibrary.wiley.com/doi/pdf/10.1002/wcms.1360 (accessed on 29 January 2024). [CrossRef]
- Vilela Oliveira, D.; Laun, J.; Peintinger, M.F.; Bredow, T. BSSE-correction scheme for consistent gaussian basis sets of double-and triple-zeta valence with polarization quality for solid-state calculations. J. Comput. Chem. 2019, 40, 2364–2376. [Google Scholar] [CrossRef]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.J.; et al. CRYSTAL 2017 User’s Manual; University of Torino: Torino, Italy, 2018. [Google Scholar]
- Mallia, G.; Orlando, R.; Roetti, C.; Ugliengo, P.; Dovesi, R. F center in LiF: A quantum mechanical ab initio investigation of the hyperfine interaction between the unpaired electron at the vacancy and its first seven neighbors. Phys. Rev. B 2001, 63, 235102. [Google Scholar] [CrossRef]
- Pascale, F.; Zicovich-Wilson, C.M.; Gejo, F.L.; Civalleri, B.; Orlando, R.; Dovesi, R. The Calculation of the Vibrational Frequencies of the Crystalline Compounds and its Implementation in the CRYSTAL Code. J. Comput. Chem. 2004, 25, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Zicovich-Wilson, C.M.; Pascale, F.; Roetti, C.; Saunders, V.R.; Orlando, R.; Dovesi, R. Calculation of the Vibration Frequencies of α-Quartz: The Effect of Hamiltonian and Basis Set. J. Comput. Chem. 2004, 25, 1873–1881. [Google Scholar] [CrossRef] [PubMed]
- Erba, A.; Ferrabone, M.; Orlando, R.; Dovesi, R. Accurate Dynamical Structure Factors from Ab Initio Lattice Dynamics: The case of Crystalline Silicon. J. Comput. Chem. 2013, 34, 346–354. [Google Scholar] [CrossRef]
- Baima, J.; Ferrabone, M.; Orlando, R.; Erba, A.; Dovesi, R. Thermodynamics and Phonon Dispersion of Pyrope and Grossular Silicate Garnets from Ab Initio Simulations. Phys. Chem. Miner. 2016, 43, 137–149. [Google Scholar] [CrossRef]
- Maschio, L.; Kirtman, B.; Rérat, M.; Orlando, R.; Dovesi, R. Ab Initio Analytical Raman Intensities for Periodic Systems Through a Coupled Perturbed Hartree-Fock/Kohn-Sham Method in an Atomic Orbital Basis. I. Theory. J. Chem. Phys. 2013, 139, 164101. [Google Scholar] [CrossRef]
- Maschio, L.; Kirtman, B.; Rérat, M.; Orlando, R.; Dovesi, R. Ab Initio Analytical Raman Intensities for Periodic Systems Through a Coupled Perturbed Hartree-Fock/Kohn-Sham Method in an Atomic Orbital Basis. II. Validation and Comparison with Experiments. J. Chem. Phys. 2013, 139, 164102. [Google Scholar] [CrossRef]
- Pople, J.; Krishnan, R.; Schlegel, H.; Binkley, J.S. Derivative studies in hartree-fock and møller-plesset theories. Int. J. Quantum Chem. 1979, 16, 225–241. [Google Scholar] [CrossRef]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. A direct MP2 gradient method. Chem. Phys. Lett. 1990, 166, 275–280. [Google Scholar] [CrossRef]
- Ferrero, M.; Rérat, M.; Orlando, R.; Dovesi, R. The Calculation of Static Polarizabilities of Periodic Compounds. The Implementation in the CRYSTAL Code for 1D, 2D and 3D Systems. J. Comput. Chem. 2008, 29, 1450–1459. [Google Scholar] [CrossRef]
- Ferrero, M.; Rérat, M.; Orlando, R.; Dovesi, R. Coupled Perturbed Hartree-Fock for Periodic Systems: The role of Symmetry and Related Computational Aspects. J. Chem. Phys. 2008, 128, 014110. [Google Scholar] [CrossRef] [PubMed]
- Zemann, J. Crystal structures, 2nd edition. Vol. 1 by R. W. G. Wyckoff. Acta Crystallogr. 1965, 18, 139. Available online: https://onlinelibrary.wiley.com/doi/pdf/10.1107/S0365110X65000361 (accessed on 29 January 2024). [CrossRef]
- Tomiki, T.; Miyata, T.; Tsukamoto, H. Temperature Dependence of the Fundamental Spectra of Potassium-Halides in the Schumann Ultraviolet Region (4.4 13.5 eV). J. Phys. Soc. Jpn. 1973, 35, 495–507. [Google Scholar] [CrossRef]
- Stoneham, A.M. Theory of Defects in Solids: Electronic Structure of Defects in Insulators and Semiconductors; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Itoh, N.; Stoneham, M. Materials Modification by Electronic Excitation; Cambridge University Press: Cambridge, MA, USA, 2000. [Google Scholar] [CrossRef]
- Lushchik, A.; Kirm, M.; Lushchik, C.; Vasil’chenko, E. Excitonic and electron–hole mechanisms of the creation of Frenkel defect in alkali halides. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2000, 166–167, 529–537. [Google Scholar] [CrossRef]
- Stammreich, H.; Forneris, R. The Raman frequency of liquid bromine. J. Chem. Phys. 1954, 22, 1624. [Google Scholar] [CrossRef]
- Suzuki, M.; Yokoyama, T.; Ito, M. Raman spectrum of the bromine crystal. J. Chem. Phys. 1969, 51, 1929–1931. [Google Scholar] [CrossRef]
- Vasilchenko, E.; Sarmukhanov, E.; Shunkeev, K.; Elango, A. Electronic excitations localized in KBr and KI crystals near vacancy defects of different sizes. Phys. Status Solidi (b) 1992, 174, 155–163. [Google Scholar] [CrossRef]
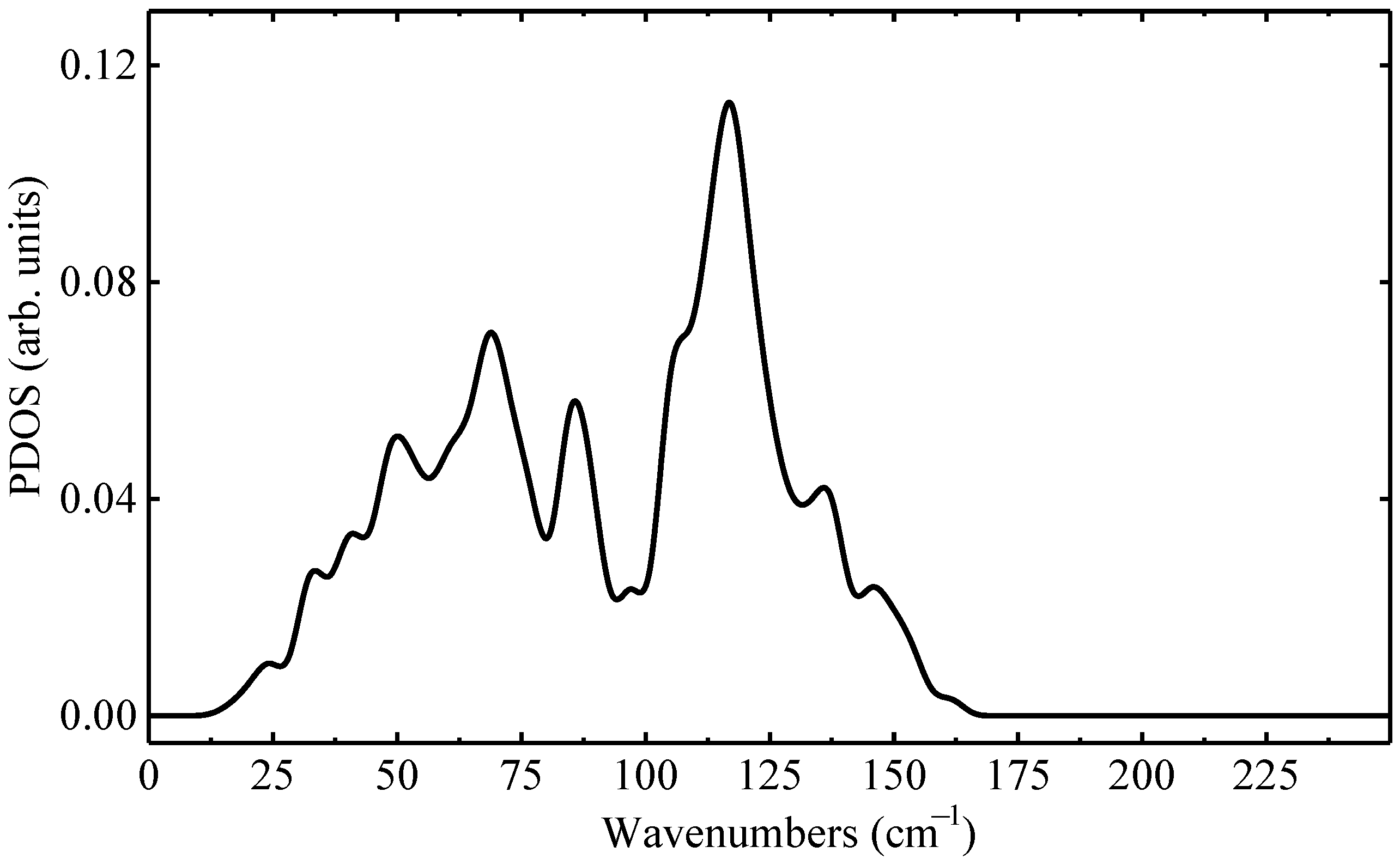

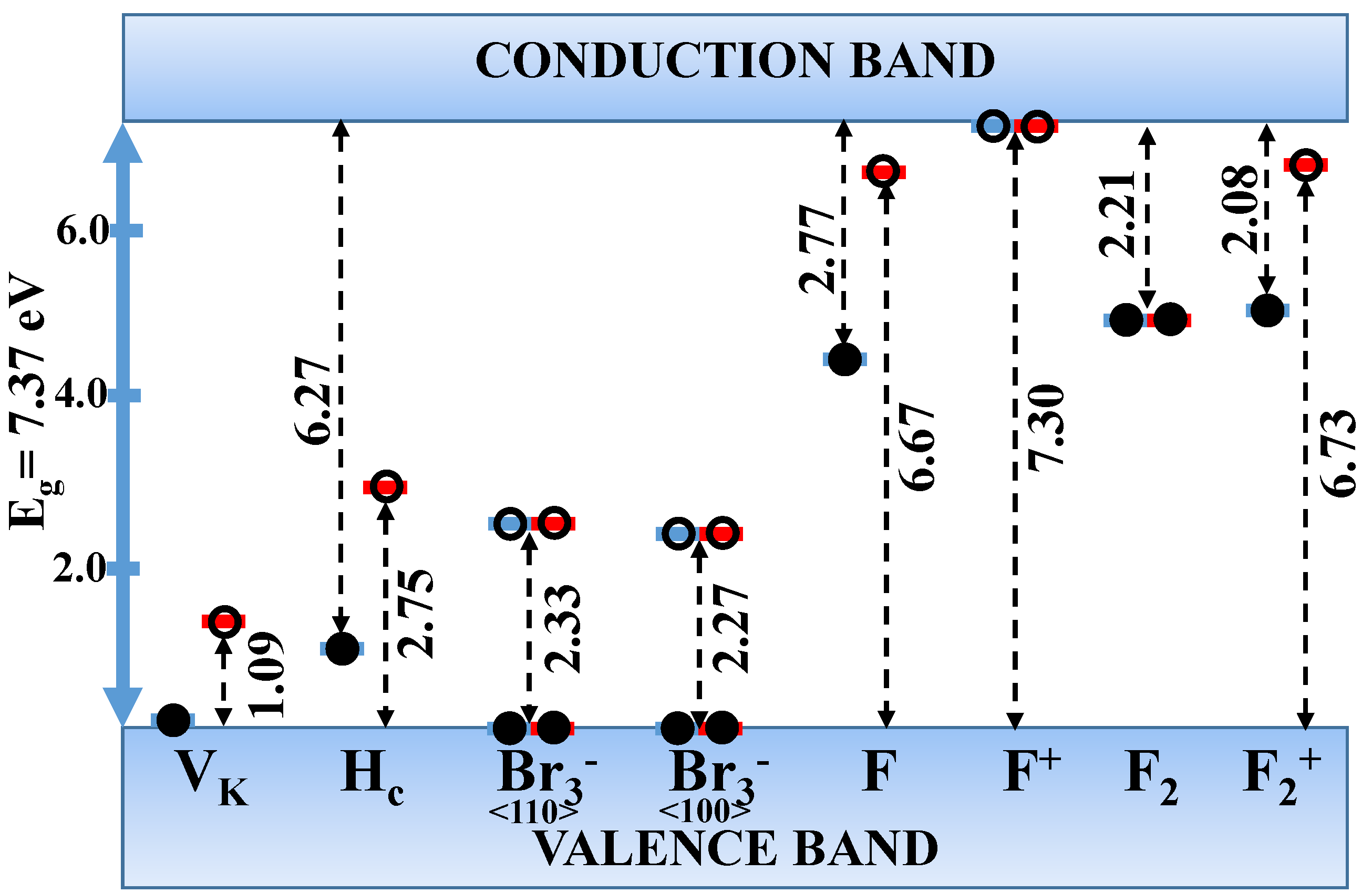

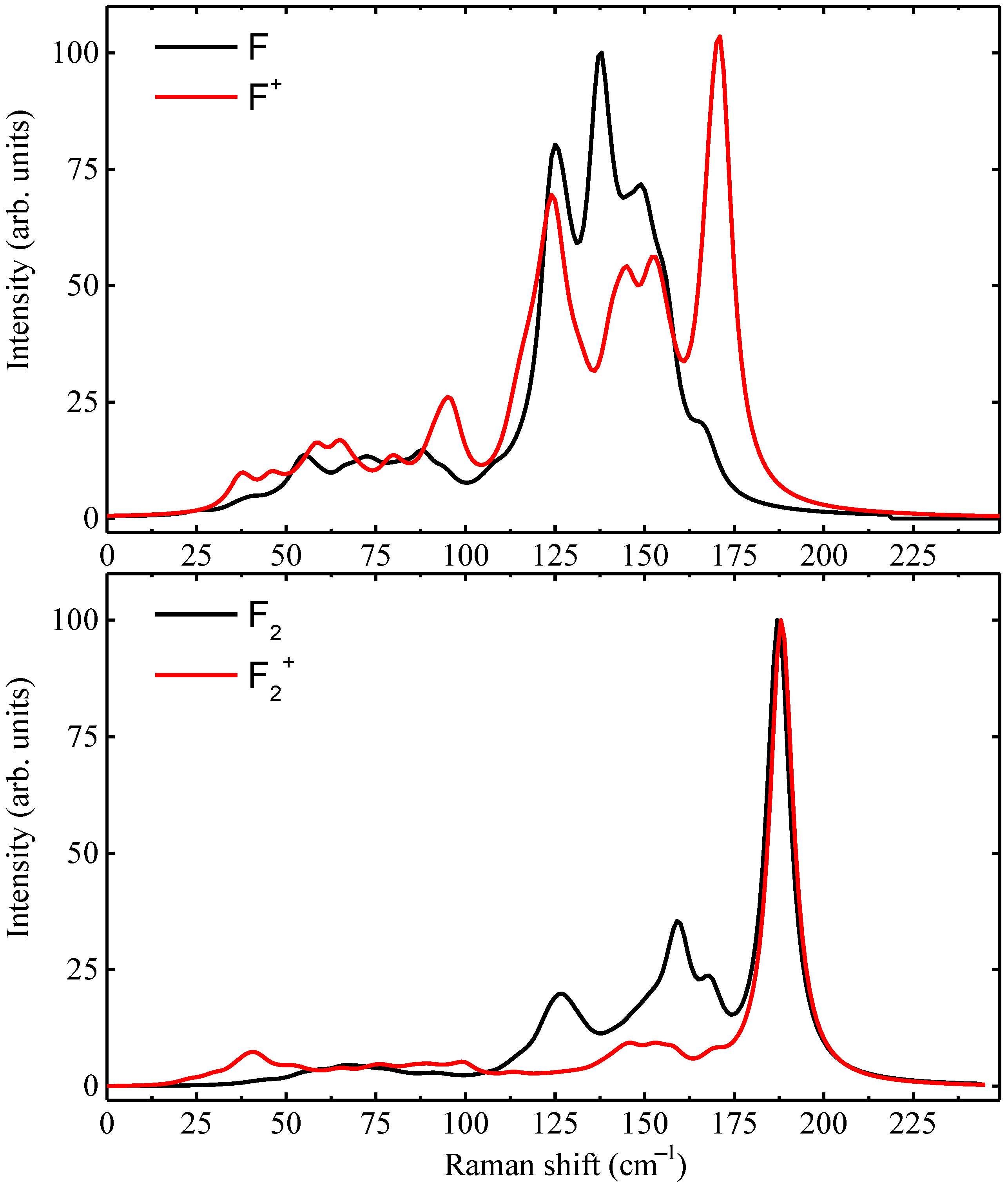
| Defect | LVM () | |||
|---|---|---|---|---|
| -center | 3.04 | −0.43/−0.43 | 0.495/0.495 | 90, 121, 165, 169, 179 |
| H-center | 2.83 | −0.46/−0.48 | 0.462/0.441 | 181, 190, 204, 207 |
| <110> | 2.59/2.65 | −0.36/−0.10/−0.43 | - | 95, 170, 173, 206, 227 |
| <100> | 2.66/2.66 | −0.46/+0.05/−0.45 | - | 68, 116, 141, 149, 186 |
| molecule | 2.38 | 0.0/0.0 | - | 302 |
| mol. ion | 2.97 | −0.5/−0.5 | 0.5/0.5 | 140 |
| mol. ion | 2.65/2.66 | −0.45/−0.10/−0.45 | - | 84, 150, 203 |
| Defect | q | LVM (cm) | |||
|---|---|---|---|---|---|
| F | 3.03 | 3.28-3.46 | −0.816 | 0.948 | 108, 124, 137, 149, 156, 167 |
| 3.57 | 3.13–3.25 | 0.0 | 0.0 | 125–135, 145, 154, 171 | |
| 2.87 / 2.53 | 3.30–3.97 | −1.598 | 0.0 | 127, 160, 169, 187 | |
| 3.53 / 3.32 | 3.15–3.35 | −0.830 | 1.028 | 153, 158, 188 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Platonenko, A.; Pankratov, V.; Kotomin, E.A.; Dauletbekova, A.; Popov, A.I. Ab Initio Study on the Vibrational and Electronic Properties of Radiation-Induced Defects in Potassium Bromide. Crystals 2024, 14, 161. https://doi.org/10.3390/cryst14020161
Platonenko A, Pankratov V, Kotomin EA, Dauletbekova A, Popov AI. Ab Initio Study on the Vibrational and Electronic Properties of Radiation-Induced Defects in Potassium Bromide. Crystals. 2024; 14(2):161. https://doi.org/10.3390/cryst14020161
Chicago/Turabian StylePlatonenko, Alexander, Vladimir Pankratov, Eugene A. Kotomin, Alma Dauletbekova, and Anatoli I. Popov. 2024. "Ab Initio Study on the Vibrational and Electronic Properties of Radiation-Induced Defects in Potassium Bromide" Crystals 14, no. 2: 161. https://doi.org/10.3390/cryst14020161